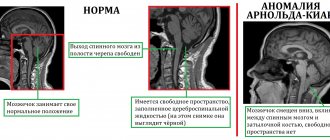
Myopathies represent a very large group of diseases with different causes, different courses and prognosis. A common feature is muscle weakness—affecting the muscles in the thighs and arms, although facial muscles may also be weakened. Sensory disorders do not occur in myopathies. Muscle weakness is usually bilateral and symmetrical from the outset, that is, similar on both sides of the body - left and right. Myopathies can be genetic or acquired. Genetically determined myopathies include, for example, muscular dystrophies, which are characterized by abnormal muscle cell structure (eg, due to a congenital deficiency of any of its components). On the other hand, acquired myopathies have inflammation (inflammatory myopathies), may accompany endocrine diseases (eg, hypothyroidism), and may also result from muscle damage by certain drugs or toxic compounds (eg, alcohol).
How common is myopathy?
The term "myopathy" is very broad and covers many different diseases. Therefore, the overall incidence of myopathy is difficult to determine. As a rule, myopathies are rare diseases. How does myopathy manifest? The main symptom of myopathy is muscle weakness. This most often affects the muscles of the thighs and arms - patients, for example, have difficulty climbing stairs, rising from a sitting position, sitting, or performing activities with raised arms (for example, hanging curtains or doing hair). Muscle weakness may be accompanied by pain. Some myopathies involve the muscles of the face or throat. There may be drooping eyelids, decreased mobility of the eyeballs, slurred speech, and difficulty swallowing. Sometimes the neck muscles are weak - then the head cannot be held up. In some myopathies, weakness of the respiratory muscles may occur, leading to shortness of breath. Sometimes with myopathies there are problems with muscle relaxation, for example, the patient cannot quickly unclench his fingers after clenching a fist. With progressive and long-term myopathies, muscle atrophy occurs. These symptoms may be accompanied by signs of damage to other tissues and organs outside the muscular system.
Classification of myopathies
To date, there is no single classification of myopathies; these diseases are divided according to several principles. The first work in this direction was the so-called clinical classification of neuromuscular diseases, which at that time were considered diseases exclusively of muscle tissue. According to this system, doctors identified such types of myopathies as limb-girdle, facioscapulohumeral, humero-tibial and others.
This division could be considered conditional, since it did not have clear characteristics, just like today. There is no more precise classification of myopathies and neuromuscular diseases in medicine, so the existing system is still used.
A pathogenetic classification is also known, the emergence of which is associated with the emergence of new knowledge about myopathy. For example, it became known that myodystrophies can manifest themselves due to multiple nerve damage, which can provoke metabolic disorders and toxic effects. Thus, myopathies began to be divided into neural amyotrophies, primary muscle diseases, etc.
With the development of medicine, pathogenetic classification is becoming more and more detailed, which is based on knowledge of the protein affected by the disease. According to this classification, the types of myopathy are calpainopathy, titinopathy, etc. Identification of a defective protein allows us to draw conclusions about the nature of its mutation.
Myopathy can be divided into hereditary and acquired types. The history of hereditary types sometimes contains relatively clear data on the presence of this disease in relatives. Examples of hereditary myopathies are dystrophic myopathies such as Duchenne myopathy, mitochondrial diseases, and storage diseases, the most common of which is Pompe disease.
Different types of myopathies have different pathogenesis, which directly depends on which gene is affected in a particular case. Thus, dystrophic myopathies or myodystrophies are a consequence of a process in which the synthesis of structural proteins of myofibrils is disrupted. Storage diseases are the result of a decrease in the amount of enzymes produced and a decrease in their activity. In this case, sooner or later the patient begins to worry about muscle weakness and muscle atrophy, which are key signs of myopathy.
How does a doctor diagnose myopathy?
The doctor takes the patient's medical history and performs a test, which usually shows paresis of the muscles (hips, arms, sometimes the face), sometimes also weakening of the reflexes caused by tapping with a neurological hammer. He then orders additional tests. Typically these are laboratory blood tests, including electrolyte, TSH, inflammation and creatine kinase levels. It is a sensitive indicator of muscle breakdown and is elevated in most patients with myopathy. It can also be increased in diseases where muscle damage is caused by nerve damage, such as amyotrophic lateral sclerosis or polyneuropathy, and sometimes in healthy people, for example after exercise or muscle injury. Electromyography (EMG) is another additional test important for diagnosis. EMG can confirm myopathy or indicate another disease that causes similar symptoms. A definitive diagnosis of myopathy, that is, determination of its type and cause, is most often possible after a muscle tissue test (for this purpose, a small piece of tissue is taken under local anesthesia for microscopic examination) or genetic analysis from a blood sample taken from the patient. Genetic testing is only available for some genetic myopathies.
Symptoms of myopathies
The main symptom indicating the presence of one of the types of myopathies is constant muscle weakness. After rest, this sensation either does not go away at all, or becomes slightly less pronounced, but still very noticeable.
The second characteristic sign is atrophy. The tissues become thin and extremely inactive. Atrophy can affect only certain muscle groups, for example, the shoulder girdle or hips, or affect all muscle tissue equally, completely depriving a person of the ability to move normally.
Extremely reduced muscle tone is also characteristic of myopathy. Each muscle in the affected area becomes flabby and flaccid to the touch. Accordingly, this affects motor activity, which cannot remain at the same level.
Due to the fact that the muscular frame ceases to perform the function of supporting the torso in the correct position, the spine begins to experience excessive loads, resulting in its curvature. The spine can curve in different ways. The most common variations are anterior to posterior curvature, resulting in lordosis or kyphosis, and a lateral curvature, as in normal scoliosis. Curvature of the spine with myopathy can become so severe that in itself it will be a significant and difficult problem to solve.
The last symptom of myopathies is the appearance of pseudohypertrophy of muscle tissue. Due to the severe thinning of some areas of the limbs, the muscles of other parts appear too large in appearance. For example, this happens with atrophy of the thigh muscles, when the calves look excessively massive against their background. In fact, in this case, the calf muscles are of normal size, but visually they look unnaturally enlarged.
How to treat myopathy?
As in the case of polyneuropathy, causal and symptomatic treatment can be distinguished. Causal treatment is possible, for example in inflammatory myopathies. In these cases, glucocorticoids (steroid hormones), immunosuppressants or intravenous immunoglobulins are used. Causal treatment of myopathy arising from endocrine diseases (eg, thyroid disease, hyperparathyroidism, or hyperadrenocorticism) involves treatment of hormonal disorders. For drug-induced or toxic myopathy, you should try to stop taking the medication or stop exposure to the toxic compound that caused the muscle damage. In rare cases of genetic myopathy, the essence of which is the absence of one of the enzymes necessary for the proper functioning of muscles, causative treatment is possible in the form of administration of the missing enzyme.
Symptomatic treatment is primarily rehabilitation. In the event that rapid muscle relaxation is a burdensome experience for a patient, pharmacological treatment is used to alleviate the symptom. Some muscle diseases affect other organs, such as the heart muscle. Then it is important for patients to undergo regular examinations with a cardiologist. In addition, genetic counseling should be given to patients with genetic myopathy.
Features of individual forms of myopathy
Research in the field of genetics has shown that individual forms of myopathy have their own characteristics of the course and clinical picture.
- According to the results, glenohumeral-facial myopathy is transmitted to descendants in an autosomal dominant manner. The first manifestations of the disease are noticeable at an early age, so if a child sleeps with his eyes open or cannot learn to whistle, parents should immediately consult a doctor. The course of the disease for a long time is not malignant, but in older age it progresses with more serious symptoms.
- The juvenile form of myopathy is transmitted only in an autosomal recessive manner. The manifestation in this case is incomplete, but this type of disease occurs most often. First, the legs and muscles of the pelvic area are affected, which almost immediately deprives the patient of the ability to walk.
- Pseudohypertrophic myopathy, which affects only boys, has a recessive mode of inheritance. Signs of myopathy appear already in the first five years of a child’s life. This is mainly atrophy, which affects almost every muscle of the legs and pelvis, combined with a mild form of debility. The progression is very rapid, leading to the early death of the patient from respiratory infections and dystrophy.
- Scapuloperoneal amyotrophy is a disease with autosomal dominant inheritance. It is characterized by a proximal distribution of atrophies in the arms and a distal distribution in the legs. In this case, in the distal parts there is a slight impairment of sensitivity.
- Distal late myopathy is transmitted by a dominant type and manifests itself at the age of 20 years. First, the muscles of the feet and arms are affected, after which the legs and forearms are involved.
- The ophthalmic form is a pathology inherited according to a dominant pattern. The disease was previously called chronic-progressive ophthalmoplegia, the cause of which was considered to be damage to nerve cells in the zone of the nuclei of the oculomotor nerves. In cases where the muscle of the tongue, palate, and larynx is affected by the disease, one should speak of a bulbar paralytic form of myopathy.
- X-chromosomal myopathy, which is benign in nature, is similar to the ophthalmological form, but proceeds more slowly, and the first symptoms appear later. Unlike other forms, patients with this type of disease can have children, and their sons are free from the defective gene, and their daughters are heterozygous carriers.
- Congenital myopathies have a recessive mode of inheritance. Their features: muscle hyponia and benign course. The possibility of having children is discussed individually for each patient by medical genetic consultation specialists.
2. Reasons
There are congenital (primary) and acquired myopathies.
Primary are caused by an inherited genetic defect, or the first genetic mutation of this kind in the family.
The essence of the defect is the incorrect coding of proteins that make up muscle tissue (cell membranes, in particular).
This leads to disturbances in the metabolism of such proteins, which entails a “chain reaction” of metabolic failures in the neuromuscular organization, gradual degeneration, dystrophy and atrophy of muscle tissue, which is manifested by a characteristic clinical picture (see below).
Secondary myopathies develop as a result of or against the background of systemic collagenosis (connective tissue diseases), inflammatory processes, endocrine disorders (for example, thyroid pathology), damage to neural tissue, and certain intoxications.
Visit our Neurology page
Treatment of Duchenne muscular dystrophy: present and future of therapy
Why is muscle function lost?
The protein dystrophin, which is absent in DMD, is necessary for the preservation of skeletal muscles. If a healthy person runs a marathon, he damages his muscles, but this is not a problem for him, because the body is able to regenerate muscle tissue. This restoration occurs in several steps. First, there is an inflammatory reaction that helps remove dead tissue from the body, and then there is a recovery stage. Since muscle damage occurs constantly in DMD, inflammation occurs continuously. A vicious inflammatory circle is formed, and muscle recovery does not occur.
In patients, the levels of oxidative stress (cell damage due to oxidation that causes people to age) are very high in their skeletal muscle, leading to fibrosis. Muscle tissue is replaced by scar or fat. Additionally, blood flow is reduced when muscles are used, further increasing oxidative stress and fibrous tissue formation. Also, patients with DMD have an excess of intracellular calcium ions, which harms the muscle power plants - mitochondria, as a result they are unable to produce energy. This is how the disease progresses and muscle function is lost.
One of the goals of DMD therapy is to combat inflammation. Reducing inflammation will not affect the disease overall, but it will slow its progression. Today, a number of drugs working in the areas listed above are being studied.
Small molecules
- Corticosteroids (prednisolone and deflazacort) suppress inflammation and slow the progression of the disease, due to which the loss of the ability to walk occurs later in patients. Corticosteroids have been used for the treatment of DMD for a long time, although they were originally developed for other diseases. There is one corticosteroid drug registered in the United States specifically for DMD. But a number of side effects are known for drugs in this group. Today, geneticists are developing safer modifications,” said Annemeke Aartsma-Rus, a professor and geneticist at the Leiden University Medical Center in the Netherlands.
— Vamorolone (a chemical modification of existing corticosteroids) is in the second stage of clinical trials in the USA and Europe. So far, the results are promising, but more research is needed.
But Edasalonexent is a drug that initially showed good results, but at the third stage of clinical trials it turned out to be less effective than corticosteroids. His research stopped.
You can also help with the disease if you reduce fibrosis. The antioxidant Idebenone was aimed at reducing oxidative stress, but in the third stage of clinical trials it was discovered that this drug had no therapeutic effect. Its development was stopped.
Pamrevlumab (anti-CTGF) is being tested in the US. This drug targets a key protein in the pathogenetic pathway. It inhibits connective tissue growth factor, which plays an important role in fibrosis.
Givinostat is also in the third stage of clinical trials. This is an epigenetic drug that changes the activity of various genes. When skeletal muscle fibers are primed for fibrosis, the body produces fewer proteins responsible for muscle tissue repair and more proteins responsible for fibrosis. Givinostat “reconfigures” and allows the production of more “correct” proteins, the expert added.
Gene replacement therapy
To resist the development of the disease, you need to learn how to add dystrophin to muscle cells. Genetic scientists are considering several methods. For example, by adding a functional copy of dystrophin to the cell. Unfortunately, it is impossible to do this by introducing healthy DNA into the body. Therefore, scientists resort to a viral vector, since viruses can introduce genetic information into cells. For this purpose, an adeno-associated virus (AAV) is used, which penetrates from the bloodstream into skeletal muscle cells. However, due to its small size, it is impossible to contain all the information about the dystrophin gene, since it is the largest in the human genome. Therefore, AAV requires microdystrophin. It includes only the most important parts of the protein and muscle-specific promoters (they begin to work only in muscles, and not in other tissues into which AAV can enter). The promoter gives the command to start the synthesis of microdystrophin. The essence of gene replacement therapy is to replace the viral genome with the microdystrophin gene and inject this virus into patients. The advantage of this therapy is that it is practically independent of the patient’s mutations. The disadvantages are the limited capacity of the virus, as well as the fact that dystrophin can be expressed in non-target organs.
— This approach works in animals (dogs and mice) and a therapeutic effect is observed. The question is how to transfer it to humans. To do this, biotechnologists need to solve the issue of scaling. The mouse weighs only 20 grams, the patient, of course, much more. Imagine that you need to prepare one liter of soup, and then 1000 liters. It will immediately become clear that you need more ingredients and a larger pan. But it won't fit on the stove. Accordingly, in order to scale, you need to change the entire production process. Of course, making soup is much easier than creating microdystrophin. Thanks to the correct choice of virus, entry of AAV into skeletal muscle was possible. It is also necessary to remember about the immune response. The body sees the adeno-associated virus, not understanding that it is useful and does not need to be touched. In addition, if the body is already familiar with this virus, then the immune response will be very intense, which is extremely dangerous. This is why preliminary screening is necessary, because if the patient has previously encountered this AAV, then this treatment method can no longer be used. Microdystrophin is expressed by skeletal muscle but is not fully functional. Over time, the cells' ability to produce microdystrophin will be lost. How quickly will this happen? It's not clear yet. But due to the immune response, we will not be able to re-treat the patient with adeno-associated virus,” noted Annemeke Aartsma-Rus.
Three companies in the world are conducting clinical trials of AAV-based drugs: Sarepta, Solid and Pfizer. Sarepta has already treated four patients under 7 years of age. They used a higher dosage of the drug and found that 80% of the muscle fibers biopsied after treatment contained about 74% microdystrophin. But minor side effects were also noted. The study was briefly suspended due to production problems, but testing was then resumed. Sarepta is currently conducting a placebo-controlled muscle study in 41 patients. Its progress and results are still unknown. Note that the FDA recommended the drug for rapid approval, i.e. Sarepta has priority. The company plans to work with older patients.
Solid treated six patients aged 4-17 years. Four dosages of the drug were used in the studies. It turned out that at low dosages less than 5% of dystrophin is produced by a small number of muscle fibers (less than 10%). 50% of muscle fibers produce microdystrophin at low levels (5 - 17%). In addition, serious side effects were observed that led to patients being hospitalized (one of the patients even had kidney failure and required short-term dialysis). Now all the patients are fine. The company suspended research twice, but it has now continued.
Pfizer is working with 13 patients aged 6-12 years. The company used half the dose of Sarepta for three patients and one and a half doses for 10 patients. It turned out that with a high dosage, dystrophin levels were restored in 50% of patients, but side effects were observed (kidney failure). All patients recovered, but this demonstrates the risks of injecting large doses of the virus. Pfizer is now preparing for a large, placebo-controlled study in 99 patients using a mid-dose.
In Russia today, two drugs are being developed that work on the basis of gene replacement therapy. One of them uses the protein utrophin. This is an autosomal analogue of the dystrophin gene and the protein performs the same functions, but in the embryonic or early period. Why do symptoms of DMD not appear immediately after birth, but only several years later? This is precisely due to the gradual replacement of utrophin with dystrophin (in patients with DMD, dystrophin cannot perform its functions).
— Utrophin was present in all patients with Duchenne muscular dystrophy. Theoretically, there should be no immune response to it, since it is a native protein for the patient. Utrophin can be used as a second line of therapy. If we take replacement gene therapy based on dystrophin, it will most likely not be possible to use it again due to the immune response. Utrophin could then be used, but using a different capsid (the outer shell of the virus) for delivery and a different serotype of AAV. For replacement therapy in mice, AAV contains a shortened version of utrophin, microutrophin. Traces of the drug are found in skeletal muscles, muscles of the diaphragm, and liver. In other tests, we see similar effects with microdystrophin,” shared the head of the Marlin Biotech gene therapy laboratory, researcher at the Laboratory of Modeling and Therapy of Hereditary Diseases of the Institute of Biogeochemistry of the Russian Academy of Sciences, molecular biologist Tatyana Egorova.
As Tatyana Egorova noted at the conference, there is an agreement between the Marlin Biotech company and the leader in the production of orphan drugs in Russia, the Generium company. Together they plan to establish the production of gene drugs based on adeno-associated viruses for clinical trials.
— To begin clinical trials, we need to conduct a series of toxicity and safety experiments on the material that will later go into clinical trials. It is impossible to talk about the time frame that will be required for this. Everything depends on the success and speed of setting up production in Russia, since, unfortunately, it does not yet exist in our country. We are completely dependent on Western supplies of equipment and reagents. In an optimistic scenario, tests will begin in our country within several years,” Tatyana Egorova shared.
Illustration prepared by the Gordey Foundation
Stop codon correction therapy
Another type of therapy being developed by foreign geneticists is reading through a stop codon. A person has a genetic code for dystrophin and a special codon (the “start” signal), which gives the body a command to begin translation. In patients with DMD, due to a point mutation in the gene, a false “stop signal” appears - a stop codon that stops protein synthesis. The result is nonfunctional dystrophin. Geneticists are working on compounds that inhibit these stops. For example, Ataluren allows you to correct the stop codon, read the entire code and produce the entire protein. This substance was conditionally approved in Europe back in 2014. There is evidence that the drug slows the progression of the disease, and it has been confirmed to be safe. But so far there is no unconditional evidence of the drug’s effectiveness.
“The European Medicines Agency has given it conditional approval, and Ataluren is already prescribed in some countries. I note that it only works on nonsense mutations (they turn the genetic code unreadable). And this is just one small group of Duchenne patients. Because the drug is commercial, the company is observing how patients fare outside of real-life trials, and indeed it is confirmed that disease progression is slowed. But an additional placebo-controlled study is required to verify the functionality of the resulting dystrophin, said Annemeke Aartsma-Rus.
Genome editing: fantasy or possibility?
A new approach in the treatment of Duchenne myodystrophin is genome editing. It has not yet undergone clinical trials, and it is too early to talk about its use in treating patients. Exon skipping and genome editing are related to the reading frame. The dystrophin gene code consists of 79 exons that are connected to each other in a certain way. So how does the CRISPR/Cas9 genome editing system work? This is a GPS system, a navigator that tells the “scissors” where to cut. With their help, a specific exon is hidden and adjacent regions are glued together, thereby restoring the reading frame of the genetic code. In the case of drug therapy, the patient needs to constantly receive medications in order to skip a certain exon. “Scissors” at the DNA level must correct the genetic code so that whenever a problem appears in the body, it would be automatically corrected.
Reading proceeds normally up to the fifth exon, but then not. Since the code is not read, protein translation stops and nonfunctional dystrophin is produced. In patients with Becker syndrome, for example, exons 48 to 51 are missing. But exon 47 can connect to 52, which means that the reading frame is not disrupted and protein translation occurs to completion. In this case, due to the absence of a gene fragment, a partially functional protein is produced. But since the important domains of the dystrophin protein are at the beginning and end of the code, the absence of the middle is not very critical. If a patient with Duchenne is missing exons 48-50, then exon 47 will not be able to connect to 51, which means the code is broken. But exon 47 could connect to 52. Accordingly, during the splicing process it would be necessary to hide exon 51 and then the gene reading frame would be restored. To do this, small modified RNAs (antisense oligonucleotides) are used in animal models. They hide a specific exon, and the result is a partially functional protein similar to that produced in patients with Becker muscular dystrophy.
“If RNA is directly introduced into the bloodstream, then after 10 seconds it will no longer be there. Chemical modification is needed to increase stability so that it can better bind to the target exon and to increase their uptake into cells. Chemical modification will prevent RNA excretion in urine. The most common are 2-o-methyl phosphorothioate and phosphorodiomidate morpholino oligomer. They are thinking about systemic delivery of antisense nucleotides. But imagine how difficult it is to do this for 700 muscles? In addition, it cannot be injected into the heart muscle. Some are talking about CRISPR cures. But this is absolutely not true. Using genome editing, you convert a Duchenne-type protein into a Becker-type protein in a patient. That is, into a partially functional protein. You are not curing the patient. With this, you can only slow down the progression of the disease,” Annemeke Aartsma-Rus said at the conference.
— The disadvantage of genome editing is that it is not suitable for all patients. For each type of mutation, its own therapy must be developed. On the plus side, for certain mutations, more domains of the protein can be preserved compared to microdystrophin. For some patients (but this is a rare pool) with exon duplications, dystrophin can be completely restored to the normal type. Another advantage is that the protein is expressed from a natural promoter. It is assumed that such therapy, once applied, will last a lifetime, unlike gene replacement therapy, Tatyana Egorova added in her report.
Experts note that the timing of intervention plays an important role in genome editing. After all, if muscle tissue has already been lost, it cannot be restored with any drugs. Therefore, for a person confined to a chair, essentially nothing will change - he will still be in the chair. Unfortunately, there are no magic wands that can turn fibrous and fatty tissue into muscle. However, drug development is underway all over the world and experts are confident that in the near future there will be a serious turn in the treatment of Duchenne muscular dystrophy.
Description
Myopathy is a disease that occurs as a result of disturbances in the metabolism and structure of muscle tissue, resulting in a decrease in the strength of the affected muscles, as well as a limitation of motor activity.
Myopathies belong to a group of neuromuscular diseases, which are characterized by dystrophic damage to muscle tissue with atrophy of individual fibers (myofibrils) in some places. In addition, atrophied myofibrils are replaced by connective or adipose tissue. This leads to the loss of the muscles’ ability to contract, which causes the appearance of muscle weakness in the clinic of the disease, as well as limitation of motor activity.
There are two main forms of the disease:
- Primary myopathies, which are often hereditary. In turn, they are divided into the following types:
- congenital myopathies that develop in infancy;
- early childhood myopathies that develop at the age of 5-10 years;
- juvenile myopathies that develop in adolescence.
- Secondary myopathies occurring against the background of other diseases.
For example, against the background of endocrine disorders (Cushing's disease, hyperaldosteronism, hyperparathyroidism, hypo- and hyperthyroidism), chronic intoxication (alcoholism, drug addiction, work in enterprises with occupational hazards), severe chronic diseases (chronic renal failure, chronic liver failure, chronic obstructive pulmonary disease, heart failure), as well as tumor processes.
Primary forms of myopathy are much more common than secondary ones. Therefore, it is extremely important when planning a pregnancy to consult a geneticist in order to identify the risk of developing this pathology in the unborn child.
Based on which part of the body muscle weakness is more pronounced, the following types of myopathy are distinguished:
- proximal - the pathological process is located in those parts of the limbs that are located as close as possible to the body (for example, in the case of the arms, these are the shoulders);
- distal - the muscles of the limbs are affected, which are at the maximum distance from the body (for example, in the case of the legs - the calf muscles);
- mixed - has symptoms of proximal and distal forms of the disease.
Primary myopathies have a poor prognosis. Especially if the first symptoms appeared in early childhood. The involvement of the heart muscle and respiratory muscles in the process is also important. In this case, the prognosis of the disease worsens. The most favorable outcome is observed with secondary myopathies, since in this case it is easier to achieve success in treating the underlying disease that caused the development of myopathy.
Diagnostic methods
Gene analysis plays a key role in determining the diagnosis. Such a study establishes the exact type of pathology. This may be Duchenne myopathy (a severe gene mutation that is fatal), Erb-Roth, Landouzy-Dejerine, etc.
Standard diagnostic measures include:
- electromyography;
- calculation of muscle enzyme activity;
- biopsy followed by histological examination to identify specific markers of myopathy.
Forecast and prevention of myopathies
The prognosis of this disease depends on the rate of development and the degree of degeneration in skeletal muscle tissue. The age at which the disease began to manifest itself also plays an important role.
The most severe case is Duchenne myopathy, the first symptoms of which occur at an early age. This type of disease very early leads to complete immobilization and causes death due to the increase in cardiac and pulmonary failure, as well as the appearance of hypoventilation and hypostatic pneumonia. Stagnation of blood in tissues can lead to infection and subsequent inflammation of the lungs.
Late forms of myopathy, the symptoms of which appear after 20-30 years, are more benign and benign in nature. The patient can lead a relatively normal life and even support himself independently, having received a profession corresponding to his level of ability to move. As a rule, this is mental and manual labor, selected depending on the level of muscle damage and does not require a high rhythm.
Preventive measures aimed at avoiding hereditary myopathies include competent family planning for couples who have relatives with this diagnosis. To do this, when thinking about a child, you should resort to medical genetic counseling with a professional assessment of the likelihood of having a child with myopathy.
Treatment of myopathies
Genetically transmitted types of myopathies cannot be cured, and therapy in this case is exclusively symptomatic. A set of measures to neutralize symptoms is selected based on the patient’s main complaints.
In most cases, the doctor prescribes orthopedic correction using special devices that give the patient the ability to move. Depending on the extent of the damage, this may include support structures or wheelchairs.
For all forms of myopathies, breathing exercises are indicated to improve lung ventilation and exercise therapy, designed to compensate for the lack of muscle load. Drug therapy includes taking thyroid hormone antagonists, which eliminate excess hormones produced by the thyroid gland.
Myopathies, caused by systemic diseases that cause disruption of collagen synthesis, are treated through the use of steroid hormones and drugs that suppress the body’s immune response.