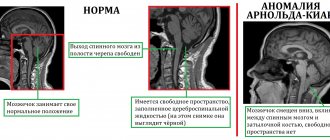
Миопатии представляют собой очень большую группу заболеваний с различными причинами, разным течением и прогнозом. Общей чертой является мышечная слабость — поражение мышц бедер и рук, хотя мышцы лица также могут быть ослаблены. Сенсорные расстройства при миопатиях не встречаются. Мышечная слабость обычно двусторонняя и симметричная с самого начала, то есть сходна с обеих сторон тела — левой и правой. Миопатии могут быть генетическими или приобретенными. Генетически обусловленные миопатии включают, например, мышечные дистрофии, которые характеризуются аномальной структурой мышечных клеток (например, из-за врожденного недостатка любого из его компонентов). С другой стороны, приобретенные миопатии имеют воспаление (воспалительные миопатии), могут сопровождать эндокринные заболевания (например, гипотиреоз), а также могут возникать в результате повреждения мышц некоторыми лекарственными средствами или токсичными соединениями (например, алкоголем).
Насколько распространена миопатия?
Термин «миопатия» очень широк и охватывает множество различных заболеваний. Поэтому общую заболеваемость миопатией трудно определить. Как правило, миопатии являются редкими заболеваниями. Как проявляется миопатия? Основным симптомом миопатии является мышечная слабость. Чаще всего это касается мышц бедер и рук — пациентам, например, трудно подниматься по лестнице, вставать из положения сидя, сидеть или выполнять действия с поднятыми руками (например, повесить шторы или делать прическу). Мышечная слабость может сопровождаться болью. При некоторых миопатиях участвуют мышцы лица или горла. Может быть опущение век, снижение подвижности глазных яблок, невнятная речь, затрудненное глотание. Иногда мышцы шеи слабые — тогда не держится голова. При некоторых миопатиях может возникнуть слабость дыхательных мышц, приводящая к одышке. Иногда при миопатиях наблюдаются проблемы с расслаблением мышц, например, пациент не может быстро разжать пальцы после сжимания кулака. При прогрессирующих и длительных миопатиях возникает атрофия мышц. Эти симптомы могут сопровождаться признаками повреждения других тканей и органов вне мышечной системы.
Классификация миопатий
На сегодняшний день единой классификации миопатий не существует, эти заболевания подразделяют по нескольким принципам. Первой работой в этом направлении была так называемая клиническая классификация нервно-мышечных заболеваний, считающихся в те времена болезнями исключительно мышечных тканей. Согласно этой системе, медики выделяли такие типы миопатий, как конечностно-поясная, лице-плече-лопаточная, гумеро-тибиальная и другие.
Данное деление можно было считать условным, так как оно не отличалось четкостью характеристик, как и сегодня. Более точной классификации миопатий и нервно-мышечных заболеваний в медицине нет, поэтому до сих пор применяется существующая система.
Известна также патогенетическая классификация, появление которой связано с возникновением новых знаний о миопатии. К примеру, стало известно, что миодистрофии могут проявляться по причине множественного поражения нервов, которые могут спровоцировать нарушение обменных процессов и токсические воздействия. Так миопатии стали подразделяться на невральные амиотрофии, первично-мышечные заболевания и др.
Все детальнее с развитием медицины становится патогенетическая классификация, в основе которой лежит знание о пораженном заболеванием белке. Согласно этой классификации видами миопатии бывают кальпаинопатия, титинопатия и пр. Выявление дефективного белка позволяет сделать выводы о характере его мутации.
Миопатия подлежит разделению на наследственный и приобретённый тип. В анамнезе наследственных типов иногда содержатся относительно четкие данные о наличие этого заболевания у родственников. Примерами миопатий наследственного типа являются дистрофические миопатии, такие, как миопатия Дюшенна, митохондриальные заболевания, а также болезни накопления, наиболее распространена из которых болезнь Помпе.
Различные виды миопатий имеют разный патогенез, что напрямую зависит от того, какой ген в конкретном случае поражен. Так дистрофические миопатии или миодистрофии являются следствием процесса, в котором происходит нарушение синтеза структурных белков миофибрилл. Болезни накопления — результат снижения количества вырабатываемых ферментов и уменьшения их активности. В данном случае рано или поздно пациента начинает беспокоить мышечная слабость и атрофия мышц, что является ключевыми признаками миопатии.
Как врач диагностирует миопатию?
Врач собирает историю болезни пациента и проводит тест, который обычно показывает парез мышц (бедра, руки, иногда лица), иногда также ослабление рефлексов, вызванных при постукивании с помощью неврологического молотка. Затем он заказывает дополнительные тесты. Обычно это лабораторные анализы крови, в том числе уровни электролита, ТТГ, воспаления и креатинкиназы. Это чувствительный показатель разрушения мышц, и его уровень повышается у большинства пациентов с миопатией. Он также может быть увеличен при заболеваниях, когда повреждение мышц вызвано повреждением нервов, таких как боковой амиотрофический склероз или полинейропатия, а иногда и у здоровых людей, например, после физической нагрузки или травмы мышц. Электромиография (ЭМГ) является еще одним дополнительным тестом, важным для диагностики. ЭМГ позволяет подтвердить миопатию или указывает на другое заболевание, которое вызывает аналогичные симптомы. Окончательный диагноз миопатии, то есть определение ее типа и причины, чаще всего возможен после теста среза мышечной ткани (для этой цели берут маленький фрагмент ткани под местной анестезией для микроскопического исследования) или генетического анализа из образца крови, взятого у пациента. Генетическое тестирование проводится только для некоторых генетических миопатий.
Симптомы миопатий
Основным симптомом, свидетельствующим о наличии одной из разновидностей миопатий, является постоянная слабость мышц. После отдыха это ощущение либо не проходит вовсе, либо становится чуть менее выраженным, но все равно весьма заметным.
Второй характерный признак — атрофия. Ткани становятся тонкими и крайне малоактивными. Атрофия может затрагивать только некоторые группы мышц, к примеру, отдел плечевого пояса или бедер или поражать все мышечные ткани в равной степени, полностью лишая человека возможности нормально двигаться.
Характерен для миопатии и крайне сниженный мышечный тонус. Каждая мышца в области поражения становится дряблой и вялой на ощупь. Соответственно, это сказывается на двигательной активности, которая не может оставаться на прежнем уровне.
Из-за того, что мышечный каркас перестает выполнять функцию поддержки туловища в правильном положении, позвоночник начинает испытывать чрезмерные нагрузки, в результате чего происходит его искривление. Позвоночник может изгибаться по-разному. Наиболее распространенные варианты — это искривление спереди назад, в результате чего развивается лордоз или кифоз, и вбок, как при обычном сколиозе. Искривление позвоночника при миопатии может принять такую серьезную степень, что уже само по себе будет являться значимой и сложно разрешимой проблемой.
Последний симптом миопатий — это появление псевдогипертрофии мышечных тканей. Из-за сильного истончения некоторых зон конечностей, мышцы других отделов внешне выглядят слишком крупными. Например, так происходит при атрофии мышц бедер, когда икры выглядят чрезмерно массивными на их фоне. На самом деле в данном случае как раз икроножные мышцы имеют нормальный размер, но визуально они выглядят неестественно увеличенными.
Как лечить миопатию?
Как и в случае с полиневропатией, можно выделить причинно-следственное и симптоматическое лечение. Возможно причинное лечение, например, при воспалительных миопатиях. В этих случаях используются глюкокортикоиды (стероидные гормоны), иммунодепрессанты или внутривенные иммуноглобулины. Причинное лечение миопатии, возникающей при эндокринных заболеваниях (например, заболевание щитовидной железы, гиперпаратиреоз или гиперадренокортицизм), включает лечение гормональных нарушений. При лекарственной или токсической миопатии нужно попытаться перестать принимать лекарство или прекратить воздействие токсического соединения, которое вызвало повреждение мышц. В редких случаях генетической миопатии, суть которой заключается в отсутствии одного из ферментов, необходимых для правильного функционирования мышц, возможно причинное лечение в виде введения недостающего фермента.
Симптоматическое лечение — это прежде всего реабилитация. В случае, если для пациента с быстрым мышечным расслаблением возникают обременительные трудности, фармакологическое лечение используется для смягчения этого симптома. При некоторых заболеваниях мышц поражаются другие органы, такие как сердечная мышца. Тогда для пациентов важно регулярно проходить осмотры у кардиолога. Кроме того, генетическое консультирование следует давать пациентам с генетической миопатией.
Особенности отдельных форм миопатии
Исследования в области генетики показали, что отдельные формы миопатии обладают своими особенностями течения и клинической картины.
- Согласно результатам, плече-лопаточно-лицевая миопатия передается потомкам по аутосомно-доминантному типу. Первые проявления заболевания заметны в раннем возрасте, поэтому если ребенок спит с открытыми глазами или не может научиться свистеть, родители должны немедленно проконсультироваться с врачом. Длительное время течение болезни не является злокачественным, но в более взрослом возрасте наступает прогресс с более серьезными симптомами.
- Ювенильная форма миопатии передается только по аутосомно-рецессивному типу. Проявляемость в данном случае неполная, но этот тип заболевания возникает наиболее часто. Вначале происходит поражение ног и мышц области таза, что практически сразу лишает больного способности ходить.
- Псевдогипертрофическая миопатия, которой страдают только мальчики, обладает рецессивным типом наследования. Признаки болезни миопатией проявляют себя уже в первые пять лет жизни ребенка. Главным образом это атрофия, которой подвергается почти каждая мышца ног и таза, сочетающаяся с легкой формой дебильности. Прогрессирование очень быстрое, приводящее к ранней смерти пациента от респираторных инфекций и дистрофии.
- Лопаточно-перонеальная амиотрофия — это заболевание с аутосомно-доминантным наследованием. Для него характерно проксимальное распределение атрофий по рукам и дистальное — по ногам. При этом в дистальных отделах наблюдается небольшое нарушение чувствительности.
- Дистальная поздняя миопатия передается по доминантном типу и проявляется в возрасте от 20 лет. Сначала происходит поражение мышц стоп и рук, после чего вовлекаются голени и предплечья.
- Офтальмологическая форма — это патология, наследуемая по доминантной схеме. Заболевание ранее называлось хронически-прогрессирующей офтальмоплегией, причиной которого считалось поражение нервных клеток в зоне ядер глазодвигательных нервов. В случаях, когда заболеванию подвергается мышца языка, неба, гортани, следует говорить о бульбарнопаралитической форме миопатии.
- Х-хромосомная миопатия, имеющая доброкачественный характер, схожа с офтальмологической формой, но протекает медленнее, а первые симптомы проявляет позже. В отличие от других форм, пациенты с данной разновидностью заболевания могут иметь детей, причем их сыновья свободны от дефектного гена, а дочери — гетерозиготные носители.
- Врожденные миопатии обладают рецессивным типом наследования. Их особенности: мышечная гипония и доброкачественное течение. Возможность рождения детей обсуждается индивидуально для каждого пациента специалистами медико-генетической консультации.
2.Причины
Различают врожденные (первичные) и приобретенные миопатии.
Первичные обусловлены наследуемым генетическим дефектом, или же первой в роду генетической мутацией такого рода.
Суть дефекта заключается в неправильной кодировке белков, входящих в состав мышечной ткани (клеточных мембран, в частности).
Это приводит к нарушениям обмена таких протеинов, что влечет за собой «цепную реакцию» метаболических сбоев в нервно-мышечной организации, постепенную дегенерацию, дистрофию и атрофию мышечной ткани, – что и проявляется характерной клинической картиной (см. ниже).
Вторичные миопатии развиваются вследствие или на фоне системных коллагенозов (заболеваний соединительной ткани), воспалительных процессов, эндокринных расстройств (например, патологии щитовидной железы), поражения нейронных тканей, некоторых интоксикаций.
Посетите нашу страницу Неврология
Лечение миодистрофии Дюшенна: настоящее и будущее терапии
Почему теряется мышечная функция?
Отсутствующий при МДД белок дистрофин необходим для сохранности скелетных мышц. Если здоровый человек бежит марафон – он повреждает мышцы, но для него это не проблема, потому что тело способно регенерировать мышечную ткань. Это восстановление происходит в несколько шагов. Сначала — воспалительная реакция, помогающая убирать из организма омертвевшие ткани, а затем – восстановительный этап. Поскольку при МДД повреждение мышц происходит постоянно, то и воспаление идет непрерывно. Формируется порочный воспалительный круг, а восстановление мышц не происходит.
У пациентов в скелетных мышцах уровень оксидативного стресса (повреждения клеток в результате окисления, из-за которого люди стареют) очень высок, это приводит к фиброзу. Мышечная ткань замещается рубцовой или жировой. Кроме того, приток крови при использовании мышц снижается, что еще больше усиливает оксидативный стресс и формирование фиброзной ткани. Также у пациентов с МДД отмечается избыток внутриклеточных ионов кальция, что вредит энергостанциям мышц – митохондриям, в результате они не могут производить энергию. Так происходит прогрессирование заболевания и потеря мышечной функции.
Одна из задач терапии МДД состоит в противостоянии воспалению. Снижение воспаления в целом не повлияет на заболевание, но замедлит его прогрессирование. Сегодня исследуется целый ряд препаратов, работающих в перечисленных выше направлениях.
Малые молекулы
— Кортикостероиды (преднизолон и дефлазакорт) подавляют воспаление и замедляют прогрессировани заболевания, благодаря этому потеря способности передвигаться у пациентов наступает позднее. При терапии МДД кортикостероиды используются давно, хотя изначально они разрабатывались для других заболеваний. В США зарегистрирован один препарат-кортикостероид, предназначенный специально для МДД. Но для препаратов этой группы известен ряд побочных эффектов. Сегодня генетики разрабатывают более безопасные модификации, — рассказала профессор, генетик медицинского центра Лейденского университета в Нидерландах Аннемеке Аартсма-Рус.
— Vamorolone (химическая модификация существующих кортикостероидов) находится на второй стадии клинических испытаний в США и Европе. Пока у него результаты многообещающие, но требуются дополнительные исследования.
А вот Edasalonexent – препарат, который изначально показывал хорошие результаты, но на третьей стадии клинических испытаний оказался менее эффективным, чем кортикостероиды. Его исследования прекратились.
Помочь при заболевании также можно, если снизить фиброз. Антиоксидант Idebenone был направлен на снижение оксидативного стресса, однако на третьей стадии клинических испытаний обнаружилось, что терапевтического эффекта у этого препарата нет. Его разработку прекратили.
В США идут испытания Pamrevlumab (anti-CTGF). Этот препарат направлен на ключевой белок в патогенетическом пути. Он тормозит фактор роста соединительной ткани, играющий важную роль в фиброзе.
На третьей стадии клинических испытаний находится и Givinostat. Это эпигенетический препарат, меняющий активность работы различных генов. Когда волокна скелетных мышц настроены на фиброз, в организме производится меньше белков, отвечающих за восстановление мышечной ткани, и больше белков, отвечающих за фиброз. Givinostat производит «перенастройку» и позволяет производить больше «правильных» белков, — добавила эксперт.
Заместительная генная терапия
Чтобы противостоять развитию заболевания, нужно научиться добавлять в мышечные клетки дистрофин. Ученые-генетики рассматривают несколько способов. Например, с помощью добавления в клетку функциональной копии дистрофина. Сделать это при помощи введения здоровой ДНК в организм, к сожалению, невозможно. Поэтому ученые прибегают к вирусному вектору, так как вирусы могут вводить в клетки генетическую информацию. Для этого используют аденоассоциированный вирус (AAV), проникающий из кровотока в клетки скелетных мышц. Однако из-за небольшого размера в него невозможно поместить всю информацию о гене дистрофина, поскольку в геноме человека он самый большой. Поэтому для AAV нужен микродистрофин. В него входят только самые важные части белка и специфичные для работы в мышцах промоторы (начинают работать только в мышцах, а не в других тканях, в которые может попасть AAV). Промотор дает команду запустить синтез микродистрофина. Суть заместительной генной терапии в том, чтобы заменить геном вируса на ген микродистрофина и данный вирус ввести пациентам. Плюс этой терапии в том, что она практически не зависит от мутаций пациента. Из минусов – ограниченная ёмкость вируса, а также то, что дистрофин может экспрессироваться в нецелевых органах.
— На животных (собаках и мышах) этот подход работает и терапевтический эффект наблюдается. Вопрос в том, как это перенести его на человека. Для этого биотехнологам необходимо решить вопрос масштабирования. Мышь весит всего 20 граммов, пациент, конечно, гораздо больше. Представьте, что вам нужно приготовить один литр супа, а затем – 1000 литров. Сразу станет понятно, что нужно больше ингредиентов и кастрюля большего размера. Но она не поместится на плите. Соответственно, чтобы масштабировать, нужно менять весь процесс производства. Конечно, сварить суп гораздо проще, чем создать микродистрофин. Благодаря правильному выбору вируса попадание AAV в скелетные мышцы стало возможным. Необходимо еще помнить об иммунном ответе. Организм видит аденоассоциированный вирус, не понимая, что он полезный и его трогать не нужно. К тому же, если организм уже знаком с этим вирусом, то иммунный ответ будет очень интенсивным, что крайне опасно. Именно поэтому нужно проводить предварительный скрининг, ведь если пациент ранее сталкивался с этим AAV, то использовать этот метод лечения уже нельзя. Микродистрофин экспрессируется скелетными мышцами, но при этом он не полностью функционален. Со временем способность клеток производить микродистрофин будет теряться. Насколько быстро это произойдет? Пока непонятно. Но из-за иммунного ответа мы не сможем повторно лечить пациента аденоассоциированным вирусом, — отметила Аннемеке Аартсма-Рус.
Три компании в мире проводят клинические испытания препаратов на основе AAV: Sarepta, Solid и Pfizer. Sarepta уже пролечили четырех пациентов в возрасте до 7 лет. Они использовали большую дозировку препарата и выяснили, что в 80% мышечных волокон, взятых при биопсии после лечения, содержалось около 74% микродистрофина. Но отмечались и незначительные побочные явления. На короткое время из-за проблем с производством исследование приостанавливалось, но затем испытания возобновили. Сейчас Sarepta проводит плацебо-контролируемое мышечное исследование на 41 пациенте. Его ход и результаты пока неизвестны. Отметим, что FDA рекомендовала препарат к быстрому одобрению, т.е. у Sarepta есть приоритет. В планах у компании – работа с пациентами более взрослого возраста.
Solid пролечили шесть пациентов в возрасте 4-17 лет. При исследованиях использовали четыре дозировки препарата. Оказалось, что при низкой дозировке производится менее 5% дистрофина небольшим количеством мышечных волокон (менее 10%). 50% мышечных волокон производят микродистрофин на низком уровне (5 — 17%). Кроме того, наблюдались серьезные побочные явления, приводившие к госпитализации пациентов (у одного из пациентов даже отказала почка, ему понадобился краткосрочный диализ). Сейчас все пациенты в порядке. Компания дважды приостанавливала исследования, но сейчас они продолжились.
Pfizer работает с 13 пациентами в возрасте 6-12 лет. Компания для трех пациентах использовала половину дозы, что вводила Sarepta, а для 10 пациентов использовала полуторную дозу. Выяснилось, что при высокой дозировке у 50% пациентов уровень дистрофина восстанавливался, но наблюдались побочные явления (отказ почек). Все пациенты восстановились, но это демонстрирует риски при введении больших доз вируса. Сейчас Pfizer готовится к большому плацебо-контролируемому исследованию на 99 пациентах с использованием средней дозировки.
В России сегодня разрабатываются два препарата, работающие на основе заместительной генной терапии. Один из них использует белок утрофин. Это аутосомный аналог гена дистрофина и белок выполняет те же функции, но в эмбриональном или раннем периоде. Почему симптомы МДД проявляются не сразу после рождения, а только спустя несколько лет? Это как раз связано с постепенной заменой утрофина на дистрофин (у пациентов с МДД дистрофин выполнять свои функции не может).
— Утрофин был у всех пациентов с миодистрофией Дюшенна. Теоретически иммунного ответа на него быть не должно, поскольку для пациента это родной белок. Утрофин может быть использован, как вторая линия терапии. Если мы возьмем заместительную генную терапию на основе дистрофина, повторно её использовать скорее всего не получится из-за иммунного ответа. Тогда можно было бы использовать утрофин, но с использованием другого капсида (внешней оболочки вируса) для доставки и другого серотипа AAV. Для заместительной терапии на мышах в составе AAV использована укороченная версия утрофина – микроутрофин. Обнаруживаются следы препарата в скелетных мышцах, мышцах диафрагмы, печени. При других тестах мы видим схожие эффекты с микродистрофином, — поделилась руководитель лаборатории генной терапии «Марлин Биотех», научный сотрудник Лаборатории моделирования и терапии наследственных заболеваний ИБГ РАН, молекулярный биолог Татьяна Егорова.
Как отметила на конференции Татьяна Егорова, существует договоренность между компанией Марлин Биотех и лидером по производству орфанных препаратов России – компанией Генериум. Совместно они планируют наладить производство генных препаратов на основе аденоассоциированных вирусов для клинических испытаний.
— Чтобы начать клинические испытания, нам нужно провести ряд экспериментов на токсичность и безопасность на том материале, который в дальнейшем пойдет в клинические испытания. Невозможно говорить о сроках, которые потребуются для этого. Все зависит от успешности и быстроты наладки производства в России, так как его в нашей стране, к сожалению, на сегодня пока не существует. Мы полностью зависим от западных поставок оборудования и реактивов. При оптимистичном раскладе испытания начнутся в нашей стране в течение нескольких лет,- поделилась Татьяна Егорова.
Иллюстрация подготовлена фондом «Гордей»
Терапия на основе исправления стоп-кодона
Еще один вид терапии, разрабатываемый зарубежными генетиками — чтение через стоп-кодон. У человека есть генетический код дистрофина и специальный кодон (сигнал «старт»), подающий организму команду начинать трансляцию. У пациентов с МДД из-за точковой мутации гена появляется ложный «стоп-сигнал» – стоп-кодон, останавливающий синтез белка. В результате получается нефункциональный дистрофин. Генетики работают над соединениями, подавляющими эти остановки. Например, Ataluren позволяет исправить стоп-кодон, считать весь код и полностью произвести белок. Это вещество условно одобрено в Европе еще в 2014 году. Есть свидетельства, что препарат замедляет прогрессирование заболевания, подтверждено, что он безопасен. Но пока нет безоговорочных доказательств эффективности препарата.
— Европейское медицинское агентство лекарственных средств дало ему условное одобрение, и в некоторых странах Ataluren уже назначается. Отмечу, что он работает только на нонсенс-мутациях (они превращают генетический код в нечитаемый). А это только одна небольшая группа пациентов с Дюшенном. Поскольку препарат коммерческий, компания наблюдает, как пациенты себя чувствуют вне испытаний в реальной жизни, и действительно, подтверждается, что прогрессирование заболевания замедляется. Но требуется еще дополнительное плацебо-контролируемое исследование, чтобы убедиться в функциональности получаемого дистрофина, — рассказала Аннемеке Аартсма-Рус.
Редактирование генома: фантастика или возможность?
Новым подходом в терапии миодистрофина Дюшенна является редактирование генома. Пока оно не проходит клинических испытаний, и говорить о его применении в лечении пациентов рано. Попуск экзона и редактирование генома связаны с рамкой считывания. Код у гена дистрофина состоит из 79 экзонов, соединяющихся друг с другом определенным образом. Так как же работает система редактирования генома CRISPR/Cas9? Это GPS-система, навигатор, который сообщает «ножницам» где резать. С их помощью прячется определенный экзон и склеиваются соседние области, благодаря чему восстанавливается рамка считывания генетического кода. В случае с медикаментозной терапией – пациенту нужно постоянно получать лекарства, чтобы пропускался определенный экзон. «Ножницы» на уровне ДНК должны исправить генетический код, чтобы всякий раз при появлении проблемы в организме, она бы автоматически исправлялась.
До пятого экзона считывание идет нормально, а потом – нет. Поскольку код не считывается, то трансляция белка останавливается и производится нефункциональный дистрофин. У пациентов с синдромом Беккера, например, отсутствуют экзоны с 48 по 51. Но 47 экзон может соединиться с 52, значит, считывающая рамка не нарушается и трансляция белка происходит до конца. При этом из-за отсутствия фрагмента гена вырабатывается частично функциональный белок. Но поскольку важные домены белка дистрофина находятся в начале и конце кода, отсутствие середины не очень критично. Если у пациента с Дюшенном отсутствуют экзоны 48-50, то 47 экзон не сможет соединиться с 51, значит, нарушается код. Но 47 экзон мог бы соединиться с 52. Соответственно, в процессе сплайсинга нужно было бы скрыть 51 экзон и тогда рамка считывания гена восстановилась бы. Для этого на животных моделях используют небольшие модифицированные RNA (антисмысловые олигонуклеотиды). Они прячут определенный экзон, и в результате получается частично функциональный белок, похожий на тот, что вырабатывается у пациентов с миодистрофией Беккера.
— Если в кровоток напрямую подать RNA, то через 10 секунд ее уже там не будет. Нужна химическая модификация для повышения стабильности, чтобы она лучше могла присоединяться к целевому экзону и чтобы повысить их усвояемость клетками. Химическая модификация позволит предотвратить выведение RNA с мочой. Самые распространенные — 2-о-метил фосфоротиоат и фосфородиомидат морфолино олигомер. Задумываются над системной доставкой антисмысловых нуклеотидов. Но представьте, как сложно это сделать для 700 мышц? Кроме того, ее невозможно ввести в мышцу сердца. Некоторые говорят о вылечивании с помощью CRISPR. Но это абсолютно не так. Вы с помощью редактирования генома превращаете у пациента белок типа Дюшенна в белок типа Беккера. То есть в частично функциональный белок. Вы не излечиваете пациента. С помощью этого вы лишь можете замедлить прогрессирование заболевания, — сообщила на конференции Аннемеке Аартсма-Рус.
— Минус редактирования генома в том, что он подойдет не для всех пациентов. Для каждого типа мутаций должна быть разработана своя терапия. Из плюсов — для определенных мутаций можно сохранить больше доменов белка по сравнению с микродистрофином. Для некоторых пациентов (но это редкий пул) с дупликацией экзонов можно полностью восстановить дистрофин до нормального типа. К плюсам относится и то, что экспрессия белка идет с естественного промотора. Предполагается, что такая терапия, единожды примененная, будет действовать всю жизнь, в отличие от заместительной генной терапии, — добавила в своем докладе Татьяна Егорова.
Эксперты отмечают, что в редактировании генома важную роль играет время вмешательства. Ведь, если мышечная ткань уже потеряна, никакими препаратами ее восстановить нельзя. Поэтому у человека, прикованного к креслу, по сути ничего не изменится – он по-прежнему будет в кресле. К сожалению, не существует волшебных палочек, способных превратить фиброзную и жировую ткани в мышечную. Однако разработка лекарственных препаратов ведется во всем мире и эксперты уверены, что в ближайшем будущем наметится серьезный поворот в терапии миодистрофии Дюшенна.
Описание
Миопатия – заболевание, которое возникает вследствие нарушения в метаболизме и строении мышечной ткани, в результате чего появляется снижение силы пораженных мышц, а также происходит ограничение двигательной активности.
Миопатии относятся к группе нервно-мышечных заболеваний, для которой характерно дистрофическое поражение мышечной ткани с атрофией отдельных волокон (миофибрилл) в некоторых местах. Кроме этого, происходит замещение атрофированных миофибрилл соединительной или жировой тканью. Это приводит к утрате мышцами способности к сокращению, что обуславливает появление в клинике заболевания мышечной слабости, а также ограничение двигательной активности.
Выделяют две основные формы заболевания:
- Первичные миопатии, которые зачастую имеют наследственный характер. В свою очередь они делятся на следующие виды:
- врожденные миопатии, которые развиваются в младенческом возрасте;
- ранние детские миопатии, которые развиваются в возрасте 5-10 лет;
- юношеские миопатии, которые развиваются в подростковом возрасте.
- Вторичные миопатии, возниающие на фоне других заболеваний.
Например, на фоне эндокринных расстройств (болезнь Иценко-Кушинга, гиперальдостеронизм, гиперпаратиреоз, гипо- и гипертиреоз), хронических интоксикаций (алкоголизм, наркомания, работа на предприятиях с профессиональными вредностями), тяжелых хронических заболеваний (хроническая почечная недостаточность, хроническая печеночная недостаточность, хроническая обструктивная болезнь легких, сердечная недостаточность), а также опухолевых процессов.
Первичные формы миопатии встречаются значительно чаще, чем вторичные. Поэтому крайне важно во время планирования беременности проконсультироваться у генетика для того, чтобы выявить риск развития данной патологии у будущего ребенка.
Исходя из того, в какой части тела мышечная слабость выражена больше, выделяют следующие виды миопатии:
- проксимальная – патологический процесс располагается в тех частях конечностей, которые максимально близко расположены к туловищу (например, в случае с руками – это плечи);
- дистальная – поражаются мышцы конечностей, которые на максимальном расстоянии находятся от туловища (например, в случае с ногами – икроножные мышцы);
- смешанная – имеет симптомы проксимальной и дистальной форм болезни.
Первичные миопатии имеют неблагоприятный прогноз. Особенно, если первые симптомы проявились в раннем детстве. Также имеет значение вовлечение в процесс сердечной мышцы и дыхательной мускулатуры. В этом случае прогноз заболевания ухудшается. Наиболее благоприятный исход наблюдается при вторичных миопатиях, так как в этом случае легче достичь успех в лечении основного заболевания, вызвавшего развитие миопатии.
Методы диагностики
Ключевое значение в определении диагноза принадлежит генному анализу. Такое исследование устанавливает точный вид патологии. Это может быть миопатия Дюшена (тяжелая генная мутация, которая заканчивается летальным исходом), Эрба-Рота, Ландузи-Дежерина и др.
К стандартным диагностическим мероприятиям относятся:
- электромиография;
- вычисление активности мышечных ферментов;
- биопсия с последующим гистологическим исследованием на предмет выявления специфических маркеров миопатии.
Прогноз и профилактика миопатий
Прогноз данного заболевания зависит от скорости развития и степени дистрофии в тканях скелетной мускулатуры. Важную роль играет и возраст, в котором болезнь начала проявлять себя.
Наиболее тяжелый случай — миопатия Дюшенна, первые симптомы которой имеют место уже в раннем возрасте. Эта разновидность заболевания очень рано приводит к полному обездвиживанию и становится причиной летального исхода ввиду нарастания сердечной и лёгочной недостаточности, а также появления гиповентиляционной и гипостатической пневмонии. Застой крови в тканях может проводить к инфицированию и последующему воспалению легких.
Поздние формы миопатии, симптомы которых проявляются после 20-30 лет, имеют более щадящий, доброкачественный характер. Пациент может вести относительно нормальный образ жизни и даже обеспечивать себя самостоятельно, получив профессию, соответствующую уровню его возможности двигаться. Как правило, это умственный и ручной труд, выбираемый в зависимости от уровня поражения мышц и не требующий высокого ритма.
Профилактические меры, направленные на избежание наследственных миопатий, заключаются в грамотном планировании семьи парами, у которых имеются родственники с данным диагнозом. Для этого, задумавшись о ребенке, следует прибегнуть к медико-генетическому консультированию с профессиональной оценкой вероятности рождения ребенка, больного миопатией.
Лечение миопатий
Генетически передаваемые типы миопатий не поддаются излечению, а терапия в данном случае исключительно симптоматическая. Комплекс мер по нейтрализации симптомов подбирается, исходя из основных жалоб больного.
В большинстве случаев врач назначает ортопедическую коррекцию при помощи специальных устройств, дающих пациенту возможность двигаться. В зависимости от степени поражения это могут быть поддерживающие конструкции или инвалидные кресла-коляски.
При всех формах миопатий показана дыхательная гимнастика для улучшения вентиляции легких и ЛФК, призванная восполнять дефицит мышечной нагрузки. Медикаментозная терапия включает в себя прием антагонистов тиреоидных гормонов, которые устраняют избыток гормонов, вырабатываемых щитовидной железой.
Миопатии, причинами которых стали системные заболевания, вызывающие нарушение синтеза коллагена, лечатся посредством применения стероидных гормонов и препаратов, угнетающих иммунную реакцию организма.