About other diseases starting with the letter “B”: Basilar impression, Basilar migraine, Beriberi, Bettolepsy, Amyotrophic lateral sclerosis, Alzheimer's disease, Wilson's disease, Hallerwarden-Spatz disease, Hamstorp disease, Hippel-Lindau disease, Canavan disease, Creutzfeldt disease- Jacob's disease, Lafora's disease, Machado-Joseph disease, Moya-moya disease, Morton's disease, Parkinson's disease, Pick's disease, Refsum's disease, Fahr's disease.
Hallervorden-Spatz disease is a progressive, inherited disorder caused by iron deposits in the brain. It is expressed by Parkinson's disease, a disruption in mental perception and in the state of consciousness, the manifestation of a convulsive symptom, contraction of muscle groups, and visual disturbances.
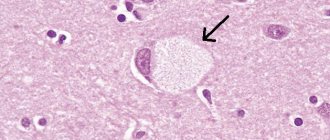
An important sign of the presence of this pathology is the “eye of the tiger” pattern in the form of a round or linear zone in the forebrain, which is visible during magnetic resonance imaging.
Therapy is aimed at eliminating the symptoms of the disease with drugs used to treat movement disorders: drugs to increase domofin levels, medications against epilepsy. Treatment is also aimed at stopping seizures and suppressing the patient’s depression. Nowadays, the disease is incurable and all actions are aimed exclusively at reducing negative manifestations.
Hallervorden-Spatz disease was characterized in 1922 by German brain research specialists, and the disease was named after the names of these morphologists. The most common clinical manifestations of this anomaly are considered to be cumulative symptoms that manifest themselves in convulsions, muscle contractions and tics. Also a distinctive feature are signs of parkinsonism, mental retardation, pathology of the optic nerves, and damage to the retina of the eye.
This pathology is quite rare. After the development of pronounced pathological phenomena after an asymptomatic course, three forms are noted: childhood, youth and adult.
Before the advent of the necessary equipment, the diagnosis was made exclusively by specialists in the field of pathological anatomy. After the introduction of the necessary equipment into medical organizations, it became possible to make a diagnosis during life. The cause of iron accumulation became known only in 2001. It turned out that the reason for everything is damage to the gene, which causes disruption of the brain. After the results of these studies, the disease was renamed in reference books to pantothenate kinase-associated neurodegeneration.
Causes of the disease
Hallervorden-Spatz disease appears due to a disruption in the functioning of genes. It can be both hereditary and accidentally acquired due to certain mutations. The genetic basis is considered to be a deviation in the pantothenate kinase gene. The genetic substrate is disturbances in the gene for photosynthesis enzymes, indicated on the map of the 20th chromosome 20 p 12.3 - p 13.
About 50 mutations have been identified. Ultimately, this genetic damage leads to a decrease in pantothenate kinase, which leads to a concentration of sulfur acid. This amino acid establishes a strong connection with iron and has a detrimental effect on proteins. As a result of the reaction, oxidation occurs, which programs cell death. In place of destroyed cells, glial cells multiply. This inexplicable reaction most often affects the paired structure of the forebrain and the substantia nigra, located in the midbrain. It is there that concentrated iron is found, which is colored brown. Also, as a result of research, formations were found that were located in the medulla, cerebral cortex and in the peripheral nervous system.
1.General information
To the synonymous diagnoses listed in the title, you can add several more names that are often used to designate this disease, for example, Osler syndrome, Randu-Osler-Weber disease, hemorrhagic telangiectasia, etc.). All of them point to the most common type of hereditary vascular pathology: its essence lies in small-focal abnormal stretching and thinning of the capillary walls, which leads to the formation of multiple telangiectasia (vascular “stars”, dots, “meshes”, etc.) with a tendency to bleeding .
The epidemiological picture is characterized by extreme unevenness and territorial dependence. Thus, Randu-Osler disease is found in every two hundred of the inhabitants of the Netherlands Antilles, while in other regions of the world the incidence may not exceed 1:100,000.
Symptoms of the disease
The most famous type of Hallervorden-Spatz disease begins in childhood between 4 and 10 years of age. The most common signs of the disease are:
Problems with muscle tone, especially in the legs. It also happens that the patient takes positions that look unnatural. In such cases, it is difficult for the child to walk.- The process then affects the torso, face and throat.
- Also with the above manifestations is dystonia, which is observed in several parts of the body. Local dystonia, involuntary spasm of the eye muscle, facial spasm, and spasmodic torticollis sometimes occur.
- Increased muscle tone, in which muscle tissue becomes dense and a state of low motor activity occurs with a limited number of movements.
- Episyndrome.
- Mental disorders such as decreased memory and alertness. Sometimes mental retardation, called oligophrenia, develops in the future. The behavior of such patients may become aggressive and deviate from moral standards.
- Impaired pronunciation and vision as a result of nerve atrophy can be identified as an independent symptom in some patients.
A diagnosis made in childhood usually leads to complete immobility after 10-15 years.
The juvenile form of Hallervorden-Spatz pathology appears at the age of 10 years and has a slow course. There are also problems with muscle tone, involuntary contractions appear, which are localized in the limbs, muscles of the mouth and jaw. There are also typical behavioral characteristics associated with intelligence and psyche.
The adult form of Hallervorden-Spatz disease is established after adulthood. It is also expressed by neurological symptoms, which are characterized by tremor and muscle tone. Insufficient motor activity, painful muscle condition, tremors and loss of coordination of movements are revealed. Loss of coordination can easily be identified using the Tavenard test - a standing patient is pushed forward, thereby throwing him off balance. The doctor is usually behind you.
The main feature of the course of the disease is considered to be a combination of neurological signs with other deviations of the muscle group. The category of impairment of mental processes can vary from the preservation of all functions to progressive dementia.
International Neurological Journal 3 (41) 2011
Hallerwarden-Spatz disease (HSD) is an inherited degenerative disease of the nervous system associated with iron accumulation in the basal ganglia [1–4, 8]. First described by German morphologists Julius Hallervorden and Hugo Spatz in 1922, GHS is considered a rare disease, although its true frequency remains unknown. Inheritance of this pathology has been established in an autosomal recessive manner. In clinical practice, the disease occurs in both sporadic and familial cases [4]. Three types have been identified depending on the age of onset of the disease: childhood, adolescence and adulthood, as well as its typical and atypical variants [10]. A significant breakthrough in knowledge regarding the mechanisms of disease development occurred in 2001, when Zhou et al. first identified a defect in the gene located on the short arm of chromosome 20, responsible for the synthesis of the enzyme pantothenate kinase, after which the disease was renamed Pantothenate Kinase associated neurodegeneration (PKAN) [5, 7, 14]. The change in the term was also partly due to publications in the literature about the participation of Hallervorden and Spatz in the “euthanasia” program for mental patients carried out by the Nazis during the war [13].
The clinical manifestations of GSH have been studied in sufficient detail over 90 years and are characterized by significant polymorphism of symptoms. The most characteristic signs of the disease are considered to be parkinsonism syndrome, various types of hyperkinesis, pyramidal signs, decreased cognitive function, pigmentary retinopathy, optic nerve atrophy, etc. [1, 2, 10]. For many years, BHS was detected only posthumously. However, due to the widespread introduction of magnetic resonance imaging (MRI) into everyday practice, the possibility of intravital diagnosis of this pathology has become possible. The characteristic MRI pattern for GSH is considered to be an oval, symmetrical hyperintense zone in the region of the globus pallidus within a larger hypointense zone. This typical symptom of GHS is called the “eye of the tiger”, and its formation is associated with extracellular iron accumulation in the basal ganglia [9, 14, 15].
In the Russian-language literature there are single publications devoted to this extremely rare pathology, which emphasize the complexity of its diagnosis in both children and adults [2, 4]. In this regard, we decided to share our own experience in diagnosing a single clinical case of late-onset GSS, accompanied by an analysis of modern literature.
Patient V., 30 years old, a permanent resident of the Republic of Belarus, was first admitted to the II Neurological Department of the 5th Clinical Hospital in Minsk in 2003 with complaints of stiffness and lack of smooth movements, impaired balance, and frequent falls during sharp turns. I was ill for 6 months when, without any apparent reason, the above-described complaints appeared and began to increase. He was treated locally for various diagnoses (most recently for multiple system atrophy) without effect. Among the past illnesses, he notes colds (rarely) and a mild traumatic brain injury in 1998. The family history is not reliably burdened. Objectively, upon admission, the general condition is satisfactory, the muscular system is well developed. Blood pressure 120/80 mm Hg, pulse 70 beats/min, rhythmic. No somatic pathology was identified. Neurologically: conscious, oriented in place and time, slightly euphoric, slightly reduced memory for current events, weakened attention. Cranial nerves without pathology, hypomimic. Strength in the limbs is sufficient, muscle tone is diffusely increased according to the extrapyramidal type. Tendon-periosteal reflexes are animated, S > D, abdominal and plantar reflexes are uniformly weakened, there are no pathological signs. Sensitivity is not impaired. Performs coordination tests satisfactorily and is stable in the Romberg position. Severe postural instability was noted. The Tavenard test is sharply positive: when it is performed, the patient falls backward without the slightest attempt to maintain balance. Autonomic tests (orthostatic, sinocarotid, oculocardiac, solar) were negative. Controls the function of the pelvic organs. The gait is devoid of smoothness, there is no physiological synkinesis; during sharp turns, the patient often loses balance and falls due to pro- and lateropulsion. During the examination: routine general clinical blood and urine tests, biochemical blood test without pathology. The content of ceruloplasmin in the blood serum is 1.7 mmol/l (normal 1.3-3.5 mmol/l), copper - 16.3 mmol/l (normal 11-22 mmol/l), iron - 15.7 mmol/l l (normal 9.5–30 mmol/l). During neuropsychological testing, the overall assessment of cognitive functions according to the Mini-Mental State Examination (MMSE) was not impaired - 27 points (norm 28–30 points). The results of the “frontal dysfunction battery” test were 14 points (norm 18 points). Consultation with an ophthalmologist: visual acuity 1.0, fundus without pathology, no Kayser-Fleischer rings detected. MRI of the brain: basal cisterns, cerebral ventricles, cortical grooves of normal size, relaxation characteristics of the brain substance are normal. In the projection of the basal ganglia on both sides, extensive symmetrical hyperintense zones with a focus of clearing (“eye of the tiger”, Fig. 1) are determined. For the purpose of differential diagnosis of the etiology of damage to the basal ganglia (Far's disease, hyperparathyroidism), a study of parathyroid hormone levels was carried out. It turned out to be within normal limits. Blood lactate and pyruvate levels were also within normal limits. He received levodopa-containing drugs, neuroprotectors, and antioxidants without significant effect. During follow-up observation for 7 years, the patient’s condition continued to slowly deteriorate; he stopped walking independently due to the inability to maintain balance. In recent years, he has been moving around using a wheelchair, his memory is slightly impaired, and movements in his limbs are preserved.
Thus, in the presented observation, the disease began at the age of 29 years, was progressive in nature and was characterized by a combination of the following neurological manifestations:
1) parkinsonism syndrome in the form of mild symmetrical hypokinesia and extrapyramidal rigidity, as well as severe postural instability, which prevailed, remaining the leading manifestation throughout the disease;
2) mild pyramidal insufficiency in the form of lateralized revitalization of the tendon-periosteal and weakening of skin reflexes;
3) mild cognitive impairment, predominantly of the subcortical type, which was manifested by a decrease in the FAB score with normal MMSE results.
The predominance of extrapyramidal syndrome in the clinical picture in a young patient justified differential diagnosis primarily with early-onset Parkinson's disease, Wilson-Konovalov disease, as well as with all forms of secondary parkinsonism and other degenerative diseases of the central nervous system.
Parkinson's disease with early onset (before 40 years of age) manifests itself as an isolated extrapyramidal syndrome, but its distinctive features are its asymmetrical nature, the frequent presence of resting tremor and, as a rule, high sensitivity to levodopa drugs, especially in the onset. Postural instability and cognitive impairment in this disease appear only with a long course, and pyramidal signs and focal changes on MRI of the brain are not typical.
Wilson-Konovalov disease (hepatocerebral degeneration) with the Slavic genotype of the disease is also characterized by onset before 30 years of age and leading neurological disorders in the form of dopa-resistant parkinsonism syndrome. However, this pathology is distinguished by frequent clinical or subclinical damage to the liver and spleen, the formation of Kayser-Fleischer rings on the iris, and typical laboratory abnormalities in the form of a decrease in serum ceruloplasmin and high copper content in the blood and urine.
It was possible to exclude all types of secondary parkinsonism (vascular, drug-induced, toxic, post-traumatic, post-encephalitic, post-anoxic, hydrocephalic) based on the lack of specific anamnestic information and the presence of characteristic changes on MRI of the brain in the form of the “eye of the tiger”.
Among other degenerative diseases, the clinical manifestations closest to the case considered were multisystem atrophy (the patient was even observed for this) and neuroacanthocytosis. However, in both cases, the disease begins at a later age (after 60 years). The cardinal distinguishing features of multisystem atrophy are the presence in the clinical picture of progressive autonomic failure and a faster rate of progression. The absence of a hereditary history indicating an autosomal dominant type of inheritance, characteristic symptoms of sensory polyneuropathy and typical hematological manifestations (acanthocytes or altered erythrocytes) allowed us to abandon the diagnosis of neuroacanthocytosis.
Thus, the combination of parkinsonism syndrome, pyramidal symptoms and neuropsychological disorders, gradual onset, progressive course, characteristic changes on MRI in the form of the “eye of the tiger” after differential diagnosis were the basis for the diagnosis of GSH, adult form, with pronounced extrapyramidal syndrome, pyramidal insufficiency , mild cognitive impairment of the subcortical type.
The etiology of the disease is unknown. Its pathogenesis is associated with multiple (more than 50) mutations of the pantothenate kinase gene (PKAN), located on the short arm of chromosome 20 at the 20p12.3-p13 locus [4]. PKAN is a regulatory enzyme for the biosynthesis of coenzyme A, which catalyzes the phosphorylation of pantothenate, N-pantothenylcysteine and panthine [15]. The pathological gene leads to a decrease in the production of this enzyme, which is accompanied by excessive accumulation of cysteine in the basal ganglia. Cysteine, in turn, binds iron ions, forming stable complexes that destroy neuronal proteins [7]. As a result, peroxidation reactions are induced with the formation of free radicals, also promoting apoptosis of basal ganglia neurons. In place of destroyed neurons, gliosis develops, giving the affected brain tissue a spongy appearance [1]. A feature of the pathogenesis of HSP is the fact that the general metabolism of iron in this disease is not impaired [5]. This is evidenced by our data on the normal level of iron in the blood serum in the presented observation. Inheritance of BHS has been established in an autosomal recessive manner, which explains the absence of a similar pathology in the patient's family among close relatives.
Two main pathomorphological markers of HDS have been identified: intense brown pigmentation and the presence of spheroid neuroaxonal formations in the basal ganglia, predominant in the area of the medial segment of the globus pallidus and the reticular part of the substantia nigra [14]. The formation of specific brown pigmentation is associated with a local high content of iron and iron-containing pigment in the intercellular space. Axonal spheroids in BHS are found not only in the basal ganglia, but also in the cortex, white matter of the cerebral hemispheres, spinal cord, and peripheral nerves. Spheroids are local extensions of presynaptic axon terminals that contain neurofilament proteins, lipofuscin granules, ubiquitin, superoxide dismutase, ferritin, and oxidized iron [6]. These pathomorphological findings allow us to classify GSH as a group of diseases called synucleinopathies, which also include Parkinson’s disease, dementia with Lewy bodies, and multiple system atrophy [5].
Clinical manifestations of GSP are characterized by polymorphism of symptoms (including intrafamilial ones), which depend on the age of onset of the disease and the type of its course [3, 11]. There are usually three clinical forms of GHS: 1) early childhood (classical) with onset at 4–10 years of age; 2) juvenile with onset at 10–18 years of age; 3) adult (atypical), developing after 18 years of age [9]. The childhood form most often manifests itself after 5 years of age with dystonia of the lower extremities (in 90%), making walking difficult. Subsequently, movement disorders usually generalize, involving the muscles of the face, pharynx, and trunk, forming various types of focal, multifocal, segmental and generalized dystonia. These may include blepharospasm, spasmodic torticollis, Meige's syndrome, writer's cramp and their combinations [11]. In 30% of cases, parkinsonism syndrome develops in the form of hypokinesia, rigidity and resting tremor [10]. Some patients have visual disorders in the form of retinal pigmentary degeneration (29%) and optic nerve atrophy (68% of patients) [9]. Characterized by mental retardation due to deterioration of memory, attention and mental disorders (aggression, antisocial behavior). This form is characterized by the fastest rate of progression with loss of the ability to move independently within 10–15 years [13].
The juvenile form is characterized by the onset of focal (limb, oromandibular) dystonia. Neuropsychological, behavioral and cognitive disorders are typical in these patients. The type of progression of symptoms of this form is slower [2, 3].
The adult form of GHS is less common (no more than 15% of all cases of the disease). It is equally manifested by various types of hyperkinesis (dystonia, choreoathetosis, hemibalism, myoclonus) and parkinsonism syndrome [2]. It has now been established that to diagnose parkinsonism syndrome, it is necessary that the patient has hypokinesia (an obligate sign), which must be accompanied by at least one of the following three symptoms: increased muscle tone of the “cogwheel” type, resting tremor and postural instability (inability to maintain balance). To assess postural instability, the doctor usually stands behind the patient and pushes him forward, towards himself and to the sides by the shoulders (Tavenard test). Normally, the patient quickly restores balance by reflexively changing the position of the feet, tilting the body, or taking one or two corrective steps. With severe parkinsonism syndrome, the patient has difficulty maintaining balance, takes several small ineffective steps forward (propulsion), backward (retropulsion), to the side (lateropulsion) or falls without any attempt to maintain balance.
In the presented observation, parkinsonism syndrome was the leading one in the clinical picture and was distinguished by a number of features. Its main manifestation was pronounced postural instability, the formation of which is explained by disruption of connections between the globus pallidus and other anatomical structures (frontal lobes, cerebellum, brainstem) [6]. At the same time, the slowness of movements with freezing and extrapyramidal rigidity described by most authors were weakly expressed in our observation, and various types of hyperkinesis were absent throughout the entire illness. The second significant feature of the extrapyramidal syndrome was its symmetry and resistance to levodopa drugs, which was also noted in the vast majority of cases of HSP described in the literature [8, 10, 11]. The reason for the latter is the preservation of the nigrostriatal dopaminergic pathway in this pathology, which is proven by the results of functional neuroimaging [2].
The pyramidal symptoms noted in our observation are a fairly typical sign of GSH, although their severity is usually much less than in the childhood form [9]. Information about the presence of cognitive deficit syndrome in patients with GHS is contradictory. On the one hand, mental and behavioral disorders in the adult form are described by researchers quite often, especially depression, aggressiveness, and emotional lability [2, 4, 6]. Their causes are associated with disruption of the frontostriatal pathways passing through the globus pallidus, which are involved in cognitive activity. At the same time, in some patients, cognitive functions remain quite intact, which was the case in the presented case. In rare cases of BHS, partial or secondary generalized seizures are observed [4]. Most patients with the adult form of GHS experience a slow course of the disease with preservation of functional activity for 15–40 years [13, 15]. However, in our observation, the rate of progression was unusually rapid. Thus, the case we presented does not fit into the framework of dividing the disease into classical and atypical forms. In the literature, the classic form of GSH is characterized by early onset, rapid progression, a typical clinical picture, and the presence of “eye of the tiger” changes on MRI. The atypical form is characterized by a late onset, slow progression, a more variable clinical picture and MRI signs in the form of the “eye of the tiger” or a hypointense zone in the area of the globus pallidus [15].
The obvious polymorphism of neurological symptoms prompted researchers to search for unified criteria for diagnosing GSP. As a result, basic and additional criteria for diagnosing GSH have been proposed. The main signs include onset in the first two decades of life, a progressive course, the presence of extrapyramidal disorders (dystonia, rigidity or choreoathetosis), a decrease in the intensity of the MRI signal from the basal ganglia (especially the globus pallidus and substantia nigra). Additional symptoms of the disease are pyramidal signs (spasticity and pathological foot reflexes), progressive decline in intelligence, retinitis pigmentosa, atrophy of the optic nerves, epileptic seizures, and a positive family history indicating an autosomal recessive type of inheritance [9]. The criteria for excluding HSP are signs of other diseases that can explain this clinical picture: hepatocerebral degeneration, Huntington's disease, and other autosomal dominant diseases. Despite obvious progress in defining the definitions of HNS, some of the mentioned criteria are not applicable to some cases, in particular, the age of onset of the disease. This is due to the fact that in the literature there are descriptions of cases of the onset of BHS in patients even older than 30 years, whose diagnosis was confirmed using DNA diagnostics [14].
A significant role in the diagnosis of GSH is assigned to the results of neuroimaging, in particular MRI brain tomograms obtained using high-field devices with a magnetic field density of at least 1–1.5 Tesla. On T2-weighted images of this pathology, 100% of typical variants of GHS reveal an oval, symmetrical hyperintense zone in the anteromedial part of the internal segment of the globus pallidus (“pupil”) within a larger hypointense zone, which is called the “eye of the tiger” [8]. An increase in signal intensity in the literature is explained by gliosis and a decrease in the number of cells with low iron content; on the contrary, a decrease in signal intensity is explained by an increase in iron concentration of more than 2 mg [12]. The timing of the appearance of this typical phenomenon in HDS is debated. According to some authors, this symptom may precede the clinical manifestations of the disease or appear 2–3 years after the onset of the disease [10]. The use of positron emission tomography makes it possible to detect a regional decrease in cerebral blood flow in the frontoparietal lobes, globus pallidus and stiatum in patients with GSH [4].
To date, there is no effective treatment for HNS. Taking into account the known pathogenesis, an attempt was made to use chelate compounds, but it was not successful, since with GSH there is only a local increase in the iron content in the brain, while its metabolism as a whole does not suffer. The use of antioxidants for their possible effect on antioxidant stress also did not lead to positive results. In this regard, treatment of this pathology is only symptomatic [2]. For parkinsonism syndrome, dopamine agonists (Mirapex, Pronoran) or amantadines (Midantan, PC-Merz) can be prescribed in individual doses. In cases of hyperkinesis, atypical benzodiazepines (clonazepam), valproates (Depakine, Encorate) or Dysport injections into the affected muscles are recommended. For spasticity, muscle relaxants (mydocalm, baclofen) are used. In order to correct cognitive impairment, drugs that affect neurotransmitter metabolism (neuromidin, gliatilin) are prescribed. To correct psychotic disorders, atypical antipsychotics (clozapine) or selective serotonin reuptake inhibitors (venlafaxine) can be used. In cases of epileptic syndrome, valproate or Topamax is indicated. A promising direction in the treatment of GSP is the use of pantothenic acid (vitamin B5) and the method of deep magnetic stimulation of the globus pallidus [10, 13].
Diagnostics
Due to the manifestation of various symptoms, it is not always possible for a neurologist to establish an accurate diagnosis of Hallervorden-Spatz disease. The dominant feature is considered to be age up to 30 years, a complex of motor activity disorders, an increase in the number of symptoms, as well as the presence of a characteristic sign on MRI images.
Additional signs of this disease may include pathological reflexes, decreased mental abilities, episyndrome, blurred vision, retinal atrophy, as well as a family history of this disease.
For diagnostic purposes, additional studies are carried out, in particular, an electroencephalogram is performed on the patient. To make a diagnosis, they rely on data from the neurological status and examination, which is based on the pattern of total electrical activity of the brain.
If there is obvious deterioration in vision, the help of an ophthalmologist is required to make a diagnosis, who will conduct a study to determine visual acuity and examine the fundus of the eye with special instruments.
A geneticist determines the presence of mutations when compiling a family tree, and also performs DNA diagnostics. Also, with the help of additional PET, reduced metabolism is detected in the formation of gray matter in the subcortical part of the brain.
There are a number of other diseases with similar pathologies, but their clinical picture does not include a number of features of Hallervorden-Spatz disease. Similar diseases in terms of symptoms: Huntington's disease, Wilson's disease, Machado-Joseph disease and other diseases associated with genetic brain damage.
When making a diagnosis, doctors rely on data obtained from MRI. All patient images show a pigmented area with iron. An image with an unambiguous appearance of this disease is called “Eye of the Tiger.”
The darker tissues of this zone look like an eye, and the lighter, bright ones look like a pupil. The moment of appearance of the “Eye of the Tiger” has not yet been determined by scientists. According to some, it appears even before certain symptoms, according to others, some time after the clear manifestation of the disease.
How can modern doctors help?
There are no treatments in modern medicine that can prevent or stop Hallervorden-Spatz disease.
Therapy is aimed at alleviating and relieving the intensity of symptoms:
- For parkinsonism syndrome, dopamine agonists (Pronoran, Pramipexole, Mirapex, Piribedil) and amantadines (Simmetrel, Midantan) are prescribed. However, the syndrome is often resistant to treatment.
- To relieve hyperkinesis, valproates (Konvulex, Depakin, Encorat) and benzodiazepines (Clonazepam, Diazepam) are used.
- To relieve muscle spasticity, muscle relaxants (Mydocalm, Baclofen) are used.
- Epileptic seizures are relieved with valproate , Tomapax.
- For cognitive impairment, Gliatilin and Neuromidin .
- For the treatment of mental disorders, it is recommended to take antipsychotics (Clonazepam, Quetiapine, Rispolent), antidepressants (Dapoxetine, Citalopram, Venlafaxine).
New methods of treating the disease are also emerging. These include therapy by administering pantothenic acid, magnetic stimulation of the brain (globus pallidus).
Treatment
Causal (etiological) therapy is unknown. There have been attempts to treat the enzyme defect. Iron chelators ("traps") such as Deferoxamine have no effect, however, treatment with the iron chelator ferriprox (deferiprone®) has been attempted since 2007. In animal experiments, deep brain stimulation led to increased dystonia and hyperkinesis. Hypokinesia can be treated with levodopa, hyperkinesia with anticholinergics. However, the effect of levodopa in patients with a PANK2 gene mutation is very questionable. Baclofen or benzodiazepines are often prescribed for muscle relaxation and pain relief.