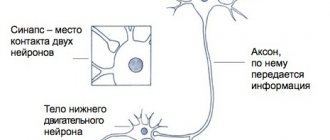
History of discovery
British ophthalmologist Warner Tay and American neurologist Bernard Sachs independently described the disease in 1887 and developed diagnostic criteria to distinguish the disease from other neurological disorders with similar symptoms.
Bernard Sachs was the first to hypothesize that this pathology is genetic. His intuitive assumption was confirmed in the mid-twentieth century after the rediscovery of Mendel's laws.
Bernard Sachs proposed a name for the new disease that can also be found in modern medical literature - amaurotic familial idiocy.
Development of the disease
A newborn suffering from Tay-Sachs syndrome looks like all children and seems quite healthy. It is common for such rare diseases to not appear immediately, and in the case of the disease in question only after six months. Up to 6 months, the child behaves in the same way as his peers. That is, he holds his head well, holds objects in his hands, makes some sounds, and perhaps begins to crawl.
Since gangliosides are not broken down in cells, enough of them accumulate for the baby to lose acquired skills. The child does not react to the people around him, his gaze is directed to one point, and apathy appears. After some period of time, blindness develops. Later, the child’s face becomes like a doll’s. Typically, children with rare diseases associated with mental retardation do not live long. In the case of Tay-Sachs disease, the baby becomes disabled and rarely lives beyond 5 years.
Symptoms of the disease in infants:
- At 3-6 months, the child begins to lose contact with the outside world. This manifests itself in the fact that he does not recognize close people, is able to react only to loud sounds, cannot focus his vision on an object, his eyes tremble, and later his vision deteriorates.
- At 10 months, the baby's activity decreases. It becomes difficult for him to move (sit, crawl, roll over). Vision and hearing become dull, apathy develops. The size of the head may increase (macrocephaly).
- After 12 months the disease gains momentum. The mental retardation of the child becomes noticeable, he very quickly begins to lose hearing, vision, muscle activity worsens, breathing difficulties arise, and seizures appear.
- At 18 months, the child is completely deprived of hearing and vision, convulsions, spastic movements, and generalized paralysis appear. The pupils do not respond to light and are dilated. Next, decerebrate rigidity develops due to brain damage.
- After 24 months, the baby suffers from bronchopneumonia and most often dies before reaching 5 years of age. If the child was able to live longer, he develops a disorder of coordination in the contractions of different muscle groups (ataxia) and a slowdown in motor skills, which progresses between 2 and 8 years.
Tay-Sachs disease also comes in other forms.
Causes of the disease
For a long time, doctors could not answer the question of what causes Tay-Sachs disease. The causes of the pathology became known only in the middle of the twentieth century, when ideas about genetics were formed. Studies have shown that the disease develops as a result of a mutation in the HEXA gene, which is located on the 15th chromosome. The disease is a type of GM2 gangliosidosis, a genetic pathology associated with a deficiency or decreased activity of hexosaminidase. Amaurotic idiocy occurs as a result of a decrease in the activity of hexazaminidase A or a deficiency of this enzyme.
The disease is transmitted in an autosomal recessive manner, therefore if a person has a healthy HEXA gene in his genotype, then he does not develop Tay-Sachs disease. The genetics of the disease is similar to the inheritance of such pathologies as Gaucher disease, Urbach-Wiethe disease, Dabin-Johnson syndrome: if both parents were carriers of the mutated gene, the probability of having a sick child is 0.25%, if both mother and father were sick, then both In children, the disease manifests itself in almost 100% of cases.
What is Tay-Sachs disease?
Tay-Sachs disease is a rare neurodegenerative disorder in which a deficiency of an enzyme (hexosaminidase A) causes the excessive accumulation of certain fats (lipids) known as gangliosides in the brain and nerve cells. This excessive accumulation of gangliosides leads to progressive dysfunction of the central nervous system. The disease belongs to the group of lysosomal storage diseases. Lysosomes are the main digestive units in cells. Enzymes in lysosomes break down or “digest” nutrients, including certain complex carbohydrates and fats. When an enzyme such as hexosaminidase A, which is needed to break down certain substances such as fats, is missing or ineffective, they accumulate in lysosomes. This is called abnormal "hoarding". When too much fat deposits accumulate in the lysosome, it becomes toxic, destroying cells and damaging surrounding tissue.
Symptoms associated with Tay-Sachs disease may include an exaggerated startle response to sudden sounds, lethargy, loss of previously acquired skills (eg, psychomotor regression), and severely decreased muscle tone (hypotonia). Infants with hypotonia may be described as “floppy.” As the disease progresses, affected infants and children may develop cherry-red spots in the middle layer of the eyes, gradual loss of vision and hearing loss, increased muscle stiffness and limited movement (spasticity), paralysis, uncontrollable electrical disturbances in the brain (epilepsy), and deterioration cognitive processes (dementia). The classic form of Tay-Sachs disease occurs in infancy. This is the most common form and is usually fatal in early childhood. There are also juvenile and adult forms of Tay-Sachs disease, but they are rare. Children with the juvenile form, also called the subacute form, develop symptoms later than children with the infantile form and usually survive into later childhood or adolescence. The adult form, also called late-onset Tay-Sachs disease, can occur any time from adolescence to the mid-30s. Symptoms and severity may vary from one person to another. Some people may be between the juvenile and adult forms.
Tay-Sachs disease is transmitted in an autosomal recessive manner. The disease results from changes (mutations) in a gene known as the HEXA gene, which regulates the production of the enzyme hexosaminidase A. The HEXA gene is found on the long arm (q) of chromosome 15 (15q23-q24). There is no cure for Tay-Sachs disease, and treatment is aimed at relieving specific symptoms that occur.
Another name for Tay-Sachs disease is GM2 gangliosidosis type 1. There are two other related diseases called Sandhoff disease and hexosaminidase activator deficiency, which are indistinguishable from Tay-Sachs disease based on symptoms and can only be differentiated by testing for underlying causes. These two disorders also cause decreased hexosaminidase activity, but are caused by changes in different genes. Collectively, these three disorders are known as GM2 gangliosidoses.
Main forms of the disease
It is customary to distinguish three main forms of the disease. The most common of them is infantile. Children with Tay-Sachs disease develop normally until 6-7 months. After this, a slow but irreversible process of decline in mental and physical abilities begins.
There is also a juvenile form of the disease. Compared to infants, it is less common. Until approximately 3-10 years of age, the child develops in the same way as his peers, but over time, a slow decline in cognitive and motor functions begins, dysarthria, dysphagia, and ataxia develop.
Late-onset Tay-Sachs disease is the rarest form of the disease. The first signs of the disease usually appear after 30 years. However, cases of earlier onset of symptoms (15-18 years) have also been recorded. This form of the disease has the most favorable prognosis, since its progression can be stopped.
Causes
Tay-Sachs disease is caused by a change (mutation) in the gene encoding the alpha subunit of hexoamindase A (HEXA). Genes provide instructions for making proteins, which play a critical role in many body functions. When a gene mutation occurs, the protein product may be faulty, ineffective, or missing. Depending on the protein's functions, it can affect many organ systems in the body, including the brain.
The HEXA gene regulates the production of the enzyme hexosaminidase A. More than 80 different mutations of the HEXA gene have been identified in people with this disease. Inheriting two mutated copies of the HEXA gene (homozygous) causes a deficiency of the enzyme hexosaminidase A, which is needed to break down a fatty substance (lipid) known as GM2 ganglioside in the body's cells. Failure to break down GM2 ganglioside leads to its abnormal accumulation in the brain and nerve cells, ultimately leading to progressive deterioration of the central nervous system.
In childhood Tay-Sachs disease, there is an almost complete absence of hexosaminidase A. In late-onset Tay-Sachs disease, there is a deficiency in the activity of the enzyme hexosaminidase A. Because of some enzyme activity, the disease is less severe and progresses much more slowly than childhood Tay-Sachs disease. The exact amount of enzyme activity in late-onset Tay-Sachs disease varies greatly from person to person. Consequently, the age of onset, severity, specific symptoms and rate of progression of late-onset disease also vary greatly from one person to another.
Changes in the HEXA gene, which causes Tay-Sachs disease, are inherited in an autosomal recessive manner. Most genetic diseases are determined by the status of two copies of a gene, one from the father and one from the mother. Recessive genetic disorders occur when a person inherits two copies of an abnormal gene for the same trait, one from each parent. If a person inherits one normal gene and one disease gene, he will be a carrier of the disease, but usually asymptomatic. The risk for two carrier parents of passing on the altered gene and having an affected child is 25% in each pregnancy. The risk of having a child who is a carrier like the parents is 50% with each pregnancy. The chance that a child will receive normal genes from both parents is 25%. The risk is the same for men and women.
Researchers have determined that the Tay-Sachs disease gene is located on the long arm (q) of chromosome 15 (15q23-q24). Chromosomes are located in the nucleus of human cells and carry the genetic information of each person. Cells in the human body usually have 46 chromosomes. Pairs of human chromosomes, numbered 1 to 22, are called autosomes, and the sex chromosomes are designated X and Y. Males have one X and one Y chromosome, and females have two X chromosomes. Each chromosome has a short arm, designated "p", and a long arm, designated "q". Chromosomes are further divided into many numbered bands. For example, "chromosome 11p13" refers to band 13 on the short arm of chromosome 11. The numbered bands indicate the location of the thousands of genes present on each chromosome.
Symptoms
Regardless of the form of the disease, several main symptoms appear: dysphagia, ataxia, loss of cognitive function, muscle atrophy. If a child under one year of age reacts sharply to sharp sounds, gains weight poorly and cannot relax his muscles, parents should show him to specialists - this is how Tay-Sachs disease begins in infants. Symptoms become increasingly severe. After 6 months, motor activity decreases, the baby loses the ability to sit independently and change position. Blindness gradually develops, hearing decreases, muscles atrophy and complete paralysis of the body develops.
In the juvenile form, in addition to the main symptoms, dysarthria (decreased speech clarity), spasticity, and impaired coordination of movements are observed. There is a gradual loss of cognitive functions - a decrease in memory, attention, and performance. Dementia develops. In the later stages of the disease, seizures appear.
The first symptoms of the adult form of the disease are difficulty swallowing, loss of coordination and dysarthria. Mental disorders similar to the symptoms of schizophrenia (visual and auditory hallucinations, apathy, decreased emotionality) often appear. Without treatment, cognitive function deteriorates. Only for this form of the disease there is an effective treatment that can stop Tay-Sachs disease. The National Guide to Neonatology says that effective methods for diagnosing the adult form of the disease appeared only in the 70s; before that, the disease was considered a childhood disease.
Symptoms of kidney failure
The kidneys not only cleanse the blood of toxins and remove them from the body, but also regulate the water-salt balance, control blood pressure levels, vascular tone, hemoglobin concentration, and help maintain the health of the musculoskeletal system and heart.
Given such a variety of organ functions, renal failure is manifested by numerous symptoms, the severity of which depends on the stage and form of the disease. In acute kidney injury at the initial stage there are only manifestations of the underlying disease. This is followed by the most severe, oliguric stage, lasting up to 3 weeks, with a decrease in daily urine volume to less than 500 ml. The first sign of the disease is a sharp decrease or cessation of urination.
The condition is often reversible (if the underlying disease or condition that led to acute renal failure is reversible) and, with proper treatment, goes into the polyuric stage of renal failure with restoration of urine volume. In this case, the patient’s well-being returns to normal, but dehydration may develop and an infection may develop. The full recovery stage lasts from six months to a year. With severe disturbances, the condition becomes chronic.
Chronic renal failure (CRF) develops slowly, over several months and even years, and leads to permanent changes in the organ. At the initial stage, there are usually no specific symptoms, but sometimes a person may notice a decrease in the volume of urine produced. Or the only manifestation of the pathology may be nocturia - frequent urination at night.
As the disease progresses and uremia develops, other symptoms appear:
- apathy, general weakness;
- thirst;
- unpleasant taste in the mouth;
- sleep disorder;
- memory impairment;
- nausea.
In the absence of effective treatment, neurological disorders occur - involuntary muscle twitching, decreased reaction speed, as well as profuse vomiting, diarrhea, shortness of breath, irritation of the skin and mucous membranes.
Establishing diagnosis
Doctors are not always able to make the correct diagnosis when it comes to such a rare pathology as Tay-Sachs disease. The symptoms, genetics and treatment of the disease are being actively studied by specialists. Regardless of the form of the disease, there are several diagnostic procedures that are performed if its presence is suspected. One of them is the determination of the activity of the enzyme hexosaminidase in blood serum, leukocytes or fibroblasts. In patients with Tay-Sachs disease, the activity of hexosaminidase B is always below normal, the enzyme hexosaminidase A is practically absent or its activity is significantly below normal.
Another important diagnostic criterion is the presence of a bright red spot on the cornea of the eye, which can be easily noticed by a therapist or ophthalmologist using an ophthalmoscope. A red spot on the cornea was found in all patients, regardless of age.
Unlike other lysosomal storage diseases (Gaucher disease, Standhoff syndrome, Niemann-Pick disease), Tay-Sachs disease does not cause enlargement of the liver and spleen (hepatosplenomegaly).
Classification of amaurotic idiocy
If we take into account the age of the child at which the first clinical manifestations began, then Tay-Sachs disease is divided into three types:
- acute infantile - occurs most often. It is characterized by the appearance of symptoms of pathology in the first months after birth. Rapidly progressing form - after two to three years the child will die;
- the late juvenile form is a rarely diagnosed species. It differs in that the first signs of pathology make themselves felt between two and ten years. As a result, a person loses previously acquired skills that are necessary for a full life: movement, speech and writing. Such patients die before the age of 15;
- chronic adult is a practically never encountered form. A peculiarity is the occurrence of symptoms of Tay-Sachs disease in adults 25-30 years old.
Treatment
There are currently no drugs to cure Tay-Sachs disease. The symptoms and treatment of the disease are still the subject of scientific research.
The infant form of Tay-Sachs disease is the most dangerous. If a sick child cannot swallow on his own, it is recommended to resort to artificial nutrition; it is impossible to restore physical skills. There are no drugs that can stop or reverse the development of the disease, despite all the efforts of scientists. Sick babies, even if they receive the best care, rarely survive past the age of four.
In the juvenile form of the disease, it is important that the child is constantly under medical supervision. Following the instructions of a specialist and undergoing all necessary medical procedures helps to extend the life of a sick child to 12-16 years.
The adult form of the disease progresses more slowly than others and is often treatable. For mental disorders, patients are prescribed lithium or cesium chloride. Clinical trials have shown that pyrimethamine can significantly slow down, and in rare cases completely stop, the progression of the disease by increasing the activity of hexosaminidase B.
Prevention of amaurotic idiocy
Prevention of the disease is possible through genetic tests. Prevention of Tay-Sachs disease is carried out in the following ways:
- prenatal diagnosis;
- preimplantation genetic testing;
- amniocentesis.
Before planning a future pregnancy, partners are advised to undergo examinations at a family planning center. If genetic testing reveals genetic defects in the 15th pair of chromosomes in both parents, they are advised not to have a child.
Also, during pregnancy, women are prescribed a number of procedures that are aimed at determining the structure of the genetic code of the unborn child.
Prenatal diagnosis
Modern research makes it possible to determine early in pregnancy whether the child has inherited the mutant HEXA gene from its parents. If both parents are carriers of the disease, it is recommended to perform a chorionic villus biopsy. This is one of the most common prenatal diagnostic procedures, the purpose of which is to identify genetic abnormalities in the fetus. It is carried out at 10-14 weeks of pregnancy. Amniocentesis also gives a clear idea of whether the child is a carrier of the mutated HEXA gene. These procedures have a risk of miscarriage of less than 1%.
In the case of artificial insemination, genetic abnormalities of the fetus can be detected even before it is implanted in the uterus. For this purpose, preimplantation genetic diagnosis is performed, an analogue of prenatal diagnosis. Its main advantage is that the procedure is non-invasive and absolutely safe. It is possible to select only healthy embryos for implantation, thereby reducing the risk of having a child with Tay-Sachs disease to almost zero.
Diagnostics
The diagnosis of Tay-Sachs disease can be confirmed by a thorough clinical assessment and specialized tests, such as blood tests that measure the level of hexosaminidase A in the body. Hexosaminidase A is reduced in patients with Tay-Sachs disease and is absent or almost absent in infants.
Molecular genetic testing can confirm the diagnosis of Tay-Sachs disease. Molecular genetic testing can detect mutations in the HEXA gene, which are known to cause the disorder but are only available as a diagnostic service in specialized laboratories.
In some cases, it is possible that the diagnosis may be suspected before birth (prenatally) based on specialized tests such as amniocentesis and chorionic villus sampling (CVS). During amniocentesis, a sample of the fluid surrounding the developing fetus is removed, while CVS involves removing tissue samples from part of the placenta. These samples are studied to determine whether hexosaminidase A is present or, as in people with Tay-Sachs disease, absent or present at significantly low levels. This is called an enzyme assay. Prenatal diagnosis is also possible through molecular genetic testing of tissue samples obtained by IVS or amniocentesis if a specific disease-causing mutation in the HEXA gene is known in the family.
Blood tests can determine whether people are carriers of the disease (that is, whether they have one copy of the disease gene). Relatives of people with Tay-Sachs disease should be tested to determine whether they are carriers of the disease gene. Couples who are planning to have a child and are of any Jewish background (not just Ashkenazi) are encouraged to undergo carrier testing before becoming pregnant.
Juvenile hexosaminidase A deficiency
This form of the disease begins to appear in children aged 2 to 5 years. The disease develops much more slowly than in infants. Therefore, the symptoms of this hereditary disease are not immediately visible. Mood swings and clumsiness in movements appear. All this does not particularly attract the attention of adults.
What happens next is:
- muscle weakness appears;
- minor cramps;
- slurred speech and impaired thought processes.
The disease at this age also leads to disability. The child lives to be 15-16 years old.