Motor neuron disease is a progressive neurological disease that destroys motor neurons, the cells that control a person's muscle activity such as walking, breathing and swallowing. Typically, signals from nerve cells in the brain (called upper motor neurons) are transmitted to and from nerve cells in the brain stem and spinal cord (called lower motor neurons) to specific muscles. Upper neurons direct lower motor neurons to perform movements such as walking or chewing. Lower motor neurons control movement in the arms, legs, torso, face, throat, and tongue. Dorsal motor neurons are also called anterior horn cells. Upper motor neurons are also called corticospanic neurons.
When disruptions occur in the transmission of signals between the lower motor neurons and a particular muscle, the muscle does not work properly; the muscles gradually weaken, and uncontrollable twitching (so-called fasciculations) may develop. When signal transmission between upper motor neurons and lower motor neurons is disrupted, limb muscles develop spasticity (stiffness), movements become slow and tense, and tendon reflexes in the knee and ankle joints become overactive. Over time, the ability to control voluntary movements may be lost.
Forms of motor neuron disease
Motor neuron diseases are classified according to whether they are inherited or sporadic, and whether the pathology is upper motor neuron or lower motor neuron dependent.
In adults, the most common form of the disease is amyotrophic lateral sclerosis (ALS), which affects both upper and lower motor neurons. The disease has hereditary and sporadic forms and can affect the arms, legs or facial muscles.
Primary lateral sclerosis is an upper motor neuron disease, whereas progressive muscular atrophy affects only the lower motor neurons in the spinal cord.
In progressive bulbar palsy, the lowest motor neurons in the brainstem are most affected, causing symptoms such as slurred speech and difficulty chewing and swallowing.
What is motor neuron disease
Motor neuron disease (MND) is a progressive neurodegenerative disease that affects motor neurons in the brain and spinal cord. The gradual death of cells in the nervous system leads to steadily increasing muscle weakness, affecting all muscle groups.
Motor neurons
The brain neurons that control movement (upper motor neurons) are located in the cerebral cortex. Their processes (axons) descend into the spinal cord, where they contact the spinal cord neuron. This contact is called a synapse. In the area of the synapse, a brain neuron releases a chemical substance (transmitter) from its process, which transmits a signal to a spinal cord neuron.
Spinal cord neurons (lower motor neurons) are located in the lower parts of the brain (bulbar) and in the cervical, thoracic, or lumbar spinal cord, depending on which muscles they send their signals to. These signals travel along the spinal cord neuron processes (axons) to the muscles and control their contractions. Bulbar neurons are responsible for contracting muscles associated with speech, chewing, and swallowing; cervical spine - for contraction of the diaphragm, arm movements; thoracic region - for body movements; lumbar region - for leg movements.
Manifestations of motor neuron damage
- When neurons in the spinal cord are damaged, muscle weakness increases, muscles lose weight (atrophy), and involuntary twitching (fasciculations) appears in them. Fasciculations don't just feel like twitching, they can also be seen. This is similar to subcutaneous muscle fluttering.
- If the neurons of the brain are affected, the muscles become weak, but at the same time stiffness (spasticity) appears, that is, muscle tone increases and it becomes difficult to relax.
- When neurons in the brain and spinal cord are simultaneously damaged, these signs can occur in different combinations. That is, muscle weakness can be accompanied by both fasciculations and muscle loss, as well as stiffness.
- Depending on which parts of the brain and spinal cord are affected, these signs may appear in the muscles responsible for the movements of the arms, legs, breathing or swallowing.
Symptoms of motor neuron diseases
Below is a brief description of the symptoms of some of the most common forms of motor neurone disease.
Amyotrophic lateral sclerosis (ALS), also called Lou Gehrig's disease or classical motor neuron disease, is a progressive, ultimately fatal disease that disrupts signaling in all muscles of the body. Many doctors use the terms motor neuron disease and ALS interchangeably. The disease is caused by disorders of both upper and lower motor neurons. The first symptoms are usually noticed in the arms and legs or in the muscles responsible for swallowing. About 75 percent of people with classic ALS develop weakness of the bulbar muscles (the muscles that control speech, swallowing and chewing). Muscle weakness and atrophy occur on both sides of the body. Patients lose strength and the ability to move their arms and legs and hold their body upright. Other symptoms include muscle spasticity, spasms, and muscle cramps. Speech may become slurred or guttural. When the muscles of the diaphragm and chest wall do not function properly, patients lose the ability to breathe without mechanical support. Although the disease generally does not impair a person's intellectual abilities or personality, some recent scientific research suggests that some people with ALS may develop cognitive (mental) impairment. Most people with amyotrophic lateral sclerosis die from respiratory failure, usually within 3 to 5 years of the onset of symptoms. However, about 10 percent of patients survive for 10 years or more.
Progressive bulbar palsy , also called progressive bulbar atrophy, affects the lower motor neurons responsible for activities such as swallowing, speaking, chewing, and others. Symptoms include weakness of the glossopharyngeal, jaw and facial muscles, progressive loss of speech function, and atrophy of the tongue muscles. Limb weakness in the disease with signs of motor neuron damage is almost always obvious, but less noticeable. People are at increased risk of choking and aspiration pneumonia caused by the passage of fluids and food through the lower respiratory tract and lungs. Affected individuals have emotional outbursts of laughing or crying (called emotional lability). Stroke and myasthenia gravis may have certain symptoms similar to those of progressive bulbar palsy and should be excluded when diagnosing this disease. In about 25 percent of people with ALS, early symptoms begin with associated bulbar abnormalities. Many clinicians believe that progressive bulbar palsy by itself, without signs of pathology in the extremities (arms or legs), is extremely rare.
Pseudobulbar palsy , which has many symptoms similar to progressive bulbar palsy, is characterized by degeneration of the upper motor neurons that transmit signals to the lower motor neurons in the brain stem. Patients develop progressive loss of the ability to speak, chew, and swallow, as well as progressive weakness of the facial muscles. Patients may develop voice disturbances and an increased gag reflex. The tongue may become immobile and lose the ability to protrude from the mouth.
Primary lateral sclerosis (PLS) damages the upper motor neurons of the arms, legs, and face. This occurs when specific nerve cells in the motor areas of the cerebral cortex (the thin layer of cells covering the brain that is responsible for most high-level brain functions) gradually degenerate, causing movements to be slow. The disease often affects the legs first, then the torso, arms, and finally the bulbar muscles. Speech may become slow and difficult. When nerve cells are damaged, leg and arm movements become clumsy, slow and weak, and spasticity occurs, resulting in the inability to walk or perform tasks that require fine hand coordination. Balance problems can lead to falls. Speech may become slow and slurred. Patients typically experience pseudobulbar affect and an overactive response. Primary lateral sclerosis is more common in men than women, and the onset of the disease usually occurs between 40 and 60 years of age. The cause of the disease is unknown. Symptoms progress gradually over many years, resulting in progressive stiffness (spasticity) and clumsiness in the affected muscles. PLS is sometimes considered a form of amyotrophic lateral sclerosis, but the main difference is the sparing of lower motor neurons, a slow rate of disease progression, and a normal life expectancy. Primary lateral sclerosis may be mistaken for spastic paraplegia, an inherited upper motor neuron disorder that causes spasms in the legs and usually begins in adolescence. Most neurologists follow the clinical course of the affected person for at least 3-4 years before making a diagnosis. This disease is not fatal, but can affect quality of life.
Progressive muscular atrophy is characterized by slow but progressive degeneration of exclusively lower motor neurons. The disease largely affects men, with onset earlier than other motor neuron diseases. Weakness usually begins in the arms and then spreads to the lower body, where it can be more severe. Other symptoms may include muscle wasting, clumsy arm movements, and muscle cramps. The muscles responsible for breathing may be damaged. Exposure to cold may worsen symptoms. The disease develops together with ALS in many cases.
Spinal muscular atrophy (SMA) is an inherited disease affecting the lower motor neurons. This is an autosomal recessive disease caused by disturbances in the SMN1 gene (the gene responsible for the production of a protein that is important for the functioning of motor neurons (SMN protein)). In SMA, insufficient levels of SMN protein lead to degeneration of lower motor neurons, causing weakness and wasting of skeletal muscles. Weakness is often more severe in the muscles of the arms and legs, as well as the muscles of the trunk. Spinal muscular atrophy in children is divided into three types based on age of onset, severity, and progression of symptoms. All three types are caused by defects in the SMN1 gene.
Post-polio syndrome (PPS) is a condition that can affect polio survivors, which can occur for decades after they have recovered from polio. Poliomyelitis is an acute viral disease that destroys motor neurons. Many people affected by the disease early in life recover, only to experience new symptoms many decades later. After acute polio, the surviving motor neurons are responsible for more of the muscles they control. Post-polio syndrome and post-polio muscle atrophy are thought to occur when surviving motor neurons are lost through aging or injury/disease. Many scientists believe that SPP is a hidden symptom of muscle weakness previously affected by polio, rather than a new motor neuron disease. Symptoms include fatigue, slowly progressive muscle weakness, muscle atrophy, cold intolerance, and muscle and joint pain. These symptoms most often occur among the muscle groups affected by the initial disease and may consist of problems breathing, swallowing, or sleeping. Other symptoms of SSP may be caused by skeletal deformities, such as long-standing scoliosis, which have led to chronic changes in the biomechanics of the joints and spine. Symptoms are more common in older people and those most affected by earlier illness. Some people have only minor symptoms, while others develop muscle wasting, which can be misdiagnosed as ALS. SSP usually does not threaten the patient’s life. According to medical statistics, 25 to 50 percent of those who have had paralytic poliomyelitis usually develop post-polio syndrome.
Diagnosis of motor neuron disease
There are no specific tests to diagnose most motor neuron diseases, although genetic testing for SMA genes currently exists. Symptoms may vary among patients and in the early stages of the disease may be similar to those of other diseases, making diagnosis difficult. The physical examination is followed by a thorough neurological examination. Neurological tests will evaluate motor and sensory skills, nervous system functioning, hearing and speech, vision, coordination and balance, mental status, and changes in mood or behavior.
Tests to rule out other diseases or measure the extent of muscle damage may include the following:
Electromyography (EMG) is used to diagnose lower motor neuron disorders, as well as muscle and peripheral nerve disorders. In an EMG, a doctor inserts a thin needle-shaped electrode attached to a recording instrument into the muscle to evaluate electrical activity during voluntary contraction and at rest. Electrical activity in the muscle is caused by lower motor neurons. When motor neurons are lost, characteristic abnormal electrical signals occur in the muscle. Testing usually takes about an hour or more, depending on the number of muscles and nerves being tested.
EMG is usually performed in combination with nerve conduction velocity testing. Nerve conduction studies measure the speed at which impulses are transmitted in nerves from small electrodes attached to the skin, as well as their strength. A small pulse of electricity (similar to a shock from static electricity) stimulates the nerve that controls a specific muscle. The second set of electrodes transmits a response electrical signal to the recording device. Nerve conduction studies help differentiate lower motor neuron disease from peripheral neuropathy and can detect abnormalities in sensory nerves.
Laboratory tests of blood, urine, and other substances can help rule out muscle diseases and other disorders that may have symptoms similar to those of motor neurone disease. For example, analysis of the cerebrospinal fluid, which surrounds the brain and spinal cord, can detect infections or inflammation that can also cause muscle stiffness. Blood tests may be ordered to measure levels of the protein creatine kinase (necessary for chemical reactions that produce energy for muscle contractions); high levels allow the diagnosis of muscle diseases such as muscular dystrophy.
Magnetic resonance imaging (MRI) uses a powerful magnetic field to produce detailed images of tissue, organs, bones, nerves and other structures of the body. MRI is often used to rule out diseases that affect the head, neck and spinal cord. MRI images can help diagnose brain and spinal cord tumors, eye diseases, inflammation, infection and vascular disorders that can lead to stroke. MRI can also detect and monitor inflammatory diseases such as multiple sclerosis. Magnetic resonance spectroscopy is a type of MRI scan that measures levels of chemicals in the brain and can be used to assess the integrity of upper motor neurons.
A biopsy of muscles or nerves can confirm the presence of neurological disorders, in particular impaired nerve regeneration. A small sample of muscle or nerve tissue is taken under local anesthetic and examined under a microscope. The sample can be removed surgically through an incision in the skin or by biopsy, in which a thin, hollow needle is inserted through the skin into the muscle. A small amount of muscle tissue remains in the hollow needle when it is removed from the body. Although this test can provide valuable information about the extent of damage, it is an invasive procedure and many experts believe that a biopsy is not always necessary for diagnosis.
Transcranial magnetic stimulation was first developed as a diagnostic tool to study areas of the brain associated with motor activity. It is also used as a treatment for certain diseases. This non-invasive procedure creates a magnetic pulse inside the brain that causes movement in an area of the body. Electrodes attached to various areas of the body collect and record electrical activity in the muscles. Measures of evoked activity can diagnose motor neuron dysfunction in motor neuron disease or monitor disease progression.
Non-motor manifestations of Parkinson's disease
S.N. Illarioshkin Doctor of Medical Sciences, Professor, Deputy Director for Research
State Scientific Center of Neurology RAMS
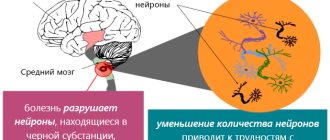
In our magazine, we have already repeatedly addressed the topic of Parkinson's disease, which is due to the high prevalence of this suffering in the population, especially in the older age group. However, this disease is so multifaceted, and the progress of recent years in studying the mechanisms of development and methods of treatment of Parkinsonism is so impressive that all new aspects of the etiology, clinical picture, diagnosis, treatment and prevention of Parkinson’s disease attract the attention of doctors, scientists and society as a whole. It is now well known that Parkinson’s disease is caused by damage to special pigmented (“black”) cells of the midbrain that synthesize the substance dopamine.
Dopamine is the main neurotransmitter of the central nervous system that regulates motor functions. A decrease in the intensity of dopamine impulse transmission in the corresponding pathways is considered as the main biochemical and neurophysiological “substrate” of Parkinson’s disease. There are three such main pathways in the brain.
The first dopamine-dependent pathway is the nigrostriatal pathway (from the Latin nigros - black and stria - stripe). This pathway, as its name suggests, leads from pigmented dopamine neurons to the striatum, a collection of motor nerve cells in the deep regions of the brain that has a characteristic striation pattern under microscopy. It is the degeneration of the nigrostriatal pathway that is responsible for the main and well-studied motor manifestations of Parkinson's disease - immobility, stiffness of various muscle groups and resting tremor.
The second dopamine pathway, also starting in the midbrain, is the mesolimbic pathway (from the Latin meso - middle). It leads to the so-called limbic system, an evolutionarily ancient structure at the base of the brain responsible for smell and emotion. Degeneration of the mesolimbic pathway in Parkinson's disease is accompanied by dysfunction of the sense of smell (a very early symptom of the disease) and various disturbances in the emotional-volitional sphere.
Among the latter, the leading place belongs to depression. Depression is considered an almost obligatory symptom of Parkinson's disease; it significantly complicates the management and care of patients and the implementation of necessary rehabilitation measures. In many cases, depression is the first manifestation of the neurodegenerative process in primary parkinsonism.
The third dopamine pathway in the brain, which also suffers from pathology of dopamine-producing cells, is the mesocortical pathway, leading to the cerebral cortex. Degeneration of this pathway is the cause of the development of impairments in cognitive functions in Parkinson's disease - memory, abstract thinking, temporal-spatial orientation, etc. In the most severe cases, which usually occur in the later stages of the disease, patients may develop dementia - a pronounced decrease in cognitive abilities, leading to social, everyday and professional maladjustment.
Other non-motor (that is, not related to the motor sphere) manifestations of Parkinson's disease are well known - autonomic disorders (constipation, sweating, greasiness of the skin), sleep disorders, low blood pressure. In general, the pathogenesis of non-motor manifestations is extremely complex and is associated not only with dopamine, but also with a number of other mediators of the nervous system. It should be emphasized that in many cases, non-motor manifestations of the disease are the leading cause of disability in patients. For example, disorders of urination, sleep and other nighttime symptoms may, according to the patients themselves, serve as the main factors in reducing the quality of life, and the onset of dementia, in addition to emerging social problems, is a contraindication for the use of surgical approaches to the treatment of the disease (surgeries on deep nuclear structures of the brain ).
Thus, timely detection of non-motor manifestations of Parkinson’s disease and their adequate, early treatment is the most important task of modern clinical neurology.
Drug treatment of Parkinson's disease involves the use of drugs of various classes that act on different “targets” of the pathological process. Thus, the “gold standard” of treatment is levodopa, a biological precursor of dopamine; Levodopa replacement therapy can compensate for the lack of dopamine in the brain and improve the motor abilities of patients. Drugs that act directly on dopamine receptors (dopamine receptor agonists) are also widely used in the clinic; An important place is occupied by drugs that affect the metabolism of dopamine and some other brain mediators. However, unfortunately, all these drugs are aimed mainly at the motor sphere and have virtually no effect on the non-motor manifestations of Parkinson’s disease and, most importantly, do not help slow down the progression of the neurodegenerative process. That is why the attention of researchers today is focused on the search for medicinal compounds that can modify the course of the disease, have a protective effect against dying neurons (neuroprotection) and at the same time help reduce the severity of non-motor symptoms resistant to treatment.
These drugs include amantadines, long-known antiparkinsonian drugs, the significance of which has recently been “rediscovered” due to the unique molecular mechanisms of action and neuroprotective (protective) properties identified. Amantadines (PC-Merz, midantan, etc.) are characterized by a complex effect: they enhance the release of dopamine into the intercellular space and slow down its reuptake, stimulate dopamine receptors, also act on other mediators (acetylcholine), etc. The drug PK-Merz (amantadine sulfate) has a number of advantages over other representatives of this class, including due to the possibility of intravenous administration and, therefore, more effective treatment of decompensations of Parkinson's disease. Amantadines have been shown to specifically block glutamate receptors, an excitatory neurotransmitter that, in excess, is seen in Parkinson's disease and contributes to neuronal damage. The toxic effect of glutamate is the most important factor in neurodegeneration, so the use of amantadine compounds can have a preventive effect and slow down the progression of the disease.
More recently, a group of researchers from Israel and Italy provided convincing evidence of the neuroprotective effect of amantadine. The researchers compared the incidence of dementia, the most important non-motor manifestation of Parkinson's disease, in patients who received and did not receive amantadine. It turned out that while taking amantadine, there was a significantly slower decline in mental abilities, and the severity of cognitive impairment in patients who used amantadine for treatment was less compared to patients who did not take amantadine. Thus, the use of amantadine helps to delay the onset of symptoms of dementia in patients with Parkinson's disease, and also reduces its severity. It is interesting to note that a similar effect in another neurodegenerative disease (Huntington's chorea) was also observed for one of the amantadine analogues, memantine.
It can be concluded that the introduction of “anti-glutamate” drugs into practice marks the beginning of a new chapter in neurology associated with influencing the mechanisms of progression of neurodegenerative diseases.
© Magazine “Nerves”, 2007, No. 1
up