Myoclonic epilepsy is a type of epileptic seizure. Characterized by a milder flow. The disease first appears in infants or young children. The onset of myoclonic epilepsy in adulthood is atypical. This form of the disease is characterized by flaccid muscle twitching. Symptoms resemble tics or hyperkinesis. In some cases, a severe course is possible. In the international classification of diseases ICD-10, myoclonic epilepsy is coded G40.3.
You can undergo diagnosis and treatment of the disease in Moscow at the Yusupov Hospital. The examination is carried out by experienced neurologists and epileptologists using modern medical equipment. The therapy meets European standards of quality and safety.
Causes of myoclonic epilepsy
At present, the exact causes of the development of myoclonic epilepsy have not been established. However, doctors identify several predisposing factors. Among them:
- Hereditary predisposition. Certain types of myoclonic epilepsy are hereditary. For example, Unferricht-Lundborg disease, Dravet syndrome. If one of your relatives has been diagnosed with epilepsy, then subsequent generations are at risk. The probability of the disease occurring is 20-30%.
- Intrauterine infection. Some infectious agents are able to penetrate the placental barrier. As a result, the risk of developing not only myoclonic epilepsy, but also mental disorders and developmental defects increases. Myoclonic epilepsy develops at the end of the 2nd-3rd trimester.
- Diseases during pregnancy are not infectious. This group of diseases includes diabetes mellitus, thyroid pathology, kidney, liver, and heart failure.
- Uncontrolled use of drugs during pregnancy. Some drugs have pronounced teratogenic properties. Therefore, their intake negatively affects the development of the fetus. Doctors advise avoiding taking medications during pregnancy. If necessary, all prescriptions must be carried out by a doctor.
- Spontaneous mutations. The reasons for such changes have not yet been studied. Provoking factors are identified, the presence of which contributes to mutation processes. These include stress, sudden changes in temperature, and excessive physical activity.
Benign myoclonic epilepsy in infants
Charlotte Dravet, Michelle Bureau
History and terminology Clinical manifestations Etiology Pathogenesis Epidemiology Prevention Differential diagnosis Diagnosis Prognosis and complications Treatment
History and terminology
Benign myoclonic epilepsy of infancy was first recognized as a distinct entity in 1981 (Dravet and Bureau 1981). Since then, quite a few cases have been described. Some authors have described cases of reflex myoclonus in response to sound or touch, proposing to classify it into 2 separate forms, calling the latter “reflex myoclonic epilepsy of infancy” (Vigevano et al 1997). We believe that this division is not appropriate and describe all cases within the framework of a single benign myoclonic epilepsy of infancy. To our knowledge, there are currently 67 published cases in the literature, of which 10 are described as reflex (Dravet and Bureau 2002). It is necessary to mention that in the first description of the syndrome, the age of onset was up to 3 years, while subsequent authors described the possibility of a later onset - up to 4 years 8 months (Giovanardi Rossi et al 1997). This means that the same type of epilepsy can appear at different times, but more often during certain periods (Guerrini et al 1994).
Clinical manifestations
Benign myoclonic epilepsy is characterized by the appearance of brief myoclonic attacks in healthy infants between ½ and 3 years of age. An earlier debut is not typical. Family history of epilepsy and febrile seizures is aggravated in 30% of children. As a rule, before the appearance of myoclonus, the child’s development proceeds normally, without signs of pathology. However, there are reports of a history of febrile seizures in approximately 20% of cases. They are usually rare, simple, and usually precede the appearance of myoclonus. Two patients had comorbidities, one had Down syndrome (Dravet and Bureau 2002) and the other had diabetes (Colamaria et al 1987). Myoclonic attacks affect the upper limbs and head, less often the lower limbs. Varying in intensity, they rarely lead to a fall. They are difficult to describe in infants; parents note head nods. They happen several times a day, unpredictably and irregularly. They are not associated with awakening, but can be triggered by a sudden sound or touch. It is quite difficult to assess the level of consciousness during an attack; with single attacks, directed activity does not stop. Only when they are grouped into clusters consisting of 2-3 repeating pseudorhythmic elements, lasting 5-10 seconds, can mild disturbances of consciousness be noted. Myoclonus can be more or less massive, involving the trunk and limbs, leading to a drop of the head and abduction of the arms outward, flexion of the lower extremities, and sometimes o or circular movements of the eyeballs. At the onset of the disease, development remains normal, and these movements are not regarded by parents and pediatricians as pathological. The interictal EEG may remain completely normal. But myoclonus is always accompanied on the EEG by fast generalized spike-wave or polyspike-wave activity, with a frequency of more than 3 Hz, more or less regular, lasting from 1 to 3 seconds. During sleep there is usually an increase in myoclonus, which usually (but not always) stops during sleep. Rhythmic photostimulation can also provoke myoclonus. Polygraphic recordings demonstrate a connection between the occurrence of myoclonus and the appearance of spike-wave or polyspike-wave discharges on the EEG. Myoclonus is short - from 1 to 3 seconds) and usually isolated. Myoclonus may be followed by a short period of atonia. Sometimes the attack may be followed by a voluntary movement that appears to be a normal muscle contraction. Interictal EEG of children is age-appropriate. Spontaneous spike-wave discharges are rarely recorded, and slow-wave activity may be observed over central areas. If rhythmic photostimulation causes spike waves, the latter are always accompanied by myoclonus. When recording sleep, its normal division into phases is recorded; during REM sleep, generalized spike-wave discharges may be noted. Children with benign myoclonic epilepsy do not develop other types of seizures, such as absence seizures or tonic seizures, even if the children do not receive treatment. Neurological status – no changes. Interictal myoclonus has been described in only 6 patients (Giovanardi Rossi et al 1997). CT or MRI examination revealed no pathological changes. The prognosis depends on the timeliness of diagnosis and treatment. If left untreated, the patient will continue to have myoclonic seizures, which can affect psychomotor development and lead to behavioral disorders. Myoclonus is easily controlled with valproate monotherapy, and the child can continue to develop in accordance with age norms.
Etiology
Benign myoclonic epilepsy is part of the group of idiopathic generalized epilepsies (Commission 1989). It may appear to be the infantile equivalent of juvenile myoclonic epilepsy, but the 2 syndromes have never been observed in the same patient. We encountered only one case (unpublished) among our patients, with onset at the age of 3 years, where the diagnosis of juvenile myoclonic epilepsy was subsequently made, but there is insufficient convincing data regarding the onset.
Pathogenesis
There is currently no clear understanding of the mechanisms of development of the syndrome. Genetics – unknown. The cases themselves are rare and no familial cases have been described. Genetic links with other forms of idiopathic generalized epilepsy have not been established. Delgado-Escueta in his study did not find a single case of juvenile myoclonic epilepsy among family members of 24 patients suffering from benign myoclonic epilepsy of infancy (Delgado-Escueta et al 1990). Arzimanoglou described the case of the second boy in the family, whose older brother suffered from a typical form of myoclonic-astatic seizures - Duse syndrome. This case raises the question of the relationship between the two syndromes within one large group of idiopathic generalized epilepsies of childhood. Biondi described a case of a patient in whom other family members were suspected of having epilepsy (Biondi et al 1991). His father and 2 sisters had short bursts of generalized spike waves recorded in their sleep EEG.
Epidemiology
According to the few epidemiological studies, benign myoclonic epilepsy of infancy accounts for less than 1% of all epilepsies (Loiseau et al 1991; Center Saint-Paul 1997, unpublished data), but about 2% of epilepsies beginning in the first 3 years of life (Dalla Bernardina et al 1983 ), and 2% of all idiopathic generalized epilepsies (Centre Saint-Paul 1997, unpublished data).
Prevention
There is no data on the possibility of preventing the syndrome.
Differential diagnosis
When myoclonus begins in the first year of life, the diagnosis that comes to mind is cryptogenic infantile spasms. Clinically, spasms differ from myoclonus. They are more intense and cause a distinct flexion of the entire body, which is never seen in benign myoclonic epilepsy. In addition to single, isolated spasms, the child always also experiences a series of spasms. Polygraphic recordings of infantile spasms show the typical pattern of short tonic contraction, well described by Fusco and Vigevano; myoclonus is rarely prolonged (Fusco and Vigevano 1993). The ictal EEG is also different - there is no fast generalized spike-wave activity. It is characterized by: a sudden flattening of the rhythm after hypsarrhythmia with (or without) the superimposition of fast-wave activity, high-amplitude slow waves followed by flattening, or even the absence of visible changes. Infantile spasms are always accompanied by a change in behavioral activity, decreased contact, impaired psychomotor development, even stopping and even regression. Interictal EEGs always record pathological changes - both true hypsarrhythmia and its modified form, or focal disorders; in this case, isolated or short bursts of bilaterally synchronous spike-waves are never recorded, as in benign myoclonic epilepsy. In cases where, after several examinations, both psychomotor development and the EEG (both during sleep and wakefulness) remain normal, even if the seizures resemble infantile spasms, then the child can be assumed to have benign nonepileptic myoclonus (Lombroso and Fejerman 1977). In these patients, there are no changes in the ictal EEG (Dravet et al 1986; Pachatz et al 1999). Severe myoclonic epilepsy of infants may also begin in the first year of life, but it always begins with prolonged, recurrent febrile seizures (but not isolated myoclonus), and psychomotor development is always impaired in this form (Dravet and Bureau 2002). If myoclonus begins after the first year of life, a diagnosis of cryptogenic Lennox-Gastaut syndrome should be considered. Seizures in this syndrome (Beaumanoir and Blume 2002) are not so much myoclonic as myoclonic-atonic, or purely atonic, and even more often tonic, which lead to sudden falls, sometimes accompanied by injuries. Their polygraphic recordings are varied, with ictal EEG characterized by an “entrainment rhythm,” or flattening of underlying activity, or a high-amplitude slow wave followed by runs of low-amplitude fast oscillations. At the beginning of the onset, the interictal EEG may remain normal, with a gradual increase in typical diffuse activity in the form of slow spike-wave complexes. The development of typical electroclinical sleep patterns may be delayed. But the diagnosis is based on a combination of different types of seizures, such as atypical absences and axial tonic seizures, persistent cognitive and mental disorders, and low effectiveness of AEDs. If myoclonic seizures remain isolated or are associated with generalized tonic-clonic seizures, a diagnosis of myoclonic-astatic epilepsy of early childhood should be considered, although the onset of myoclonic-astatic seizures in this syndrome is rarely observed before the age of 3 years (Doose 1992). There are significant differences between these two syndromes: (1) the clinical manifestations of seizures, which always lead to falls in the case of myoclonic-astatic seizures, while falls are quite rare in benign myoclonic epilepsy, as well as the combination with other types of seizures - often status petit mal seizures with stupor, which is never seen in benign myoclonic epilepsy (Guerrini et al 1994); (2) various types of EEG abnormalities. Spike waves and polyspike waves are more numerous, and are grouped into long bursts, combined with a typical theta rhythm in the central-parietal regions. But some of the cases that Doose included in this syndrome should be classified as benign myoclonic epilepsy. It is also possible that the “myoclonic epilepsy of infancy” group in the Delgado-Escueta studies (Delgado-Escueta et al 1990) included both patients with myoclonic-astatic seizures and benign myoclonic epilepsy of infancy. Finally, it is necessary to exclude other epilepsies that can debut in the first 3 years of life and manifest mainly as myoclonus and have a different prognosis. They present various combinations of other seizure types, with focal EEG changes, delayed psychomotor development, poor response to AED therapy and an uncertain prognosis (Dravet 1990).
Diagnostics
The diagnostic algorithm is quite simple. A high-quality medical history and repeated polygraphic video-EEG recordings are necessary to prove the presence of myoclonic attacks with generalized spike-wave discharges. Myoclonus is either spontaneous or occurs in response to sound, touch or rhythmic photostimulation, as well as during sleep. During EEG sleep, you can see some activation of discharges without changing their morphology, the appearance of fast rhythms and focal disturbances. Neuroimaging is useful (but not necessary) to confirm the absence of structural brain abnormalities. Neuropsychological testing is necessary to verify the absence of psychomotor developmental disorders.
Forecast
By definition, the prognosis is favorable, myoclonic seizures stop provided that appropriate treatment is prescribed - monotherapy with valproate. One study reported that only 5 patients required the addition of a second drug to achieve seizure control (Giovanardi Rossi et al 1997). Available data on follow-up duration vary, ranging from 9 months to 27 years. In 10 patients, rare generalized tonic-clonic seizures appeared, not combined with myoclonus, of which in 3 they occurred after discontinuation of the drug, in the rest - in adolescence (Dravet and Bureau 2002). Seizures triggered by sound or touch are easier to control than spontaneous ones. Conversely, photosensitivity is more difficult to control and may persist for several years after the attacks have stopped. It is more difficult to predict the outcome based on mental development. In most cases, the prognosis is quite favorable. However, longitudinal studies have described 12 patients who presented with mild mental retardation, personality disorders or mild behavioral disturbances (Colamaria et al 1987; Todt and Muller 1992; Giovanardi Rossi et al 1997; Dravet and Bureau 2002). None of these patients were hospitalized for specialized treatment. A favorable prognosis for psychological and cognitive functions also depends on the timeliness of the diagnosis, the appointment of adequate treatment and assistance from relatives. But there are also opposite factors, such as problems in the family and unfavorable characteristics of the relationship between mother and child.
Treatment
First of all, monotherapy with valproate is prescribed, preferably by injection, because children may refuse to drink syrup. It is necessary to carefully monitor its level in the blood plasma, since irregular use can lead to resumption of attacks and imitate the resistant form. A daily dosage of 30 mg/kg is usually sufficient, but sometimes it is necessary to increase the dose (Lin et al 1998). Valproate is also effective against febrile seizures. If myoclonus does not completely resolve on valproate, you can try adding a benzodiazepine (clobazam or nitrazepam) or ethosuximide, or reconsider the diagnosis. Therapy, if well tolerated, should be continued for 3-4 years from the onset of attacks, longer in cases of photosensitivity. If the attacks are purely reflexive in nature, you can do without taking valproate or stop therapy earlier. If one generalized tonic-clonic seizure occurs during adolescence, another, shorter course of treatment may be required.
Bibliography
Arzimanoglou A, Prudent M, Salefranque F. Epilepsie myoclono-astatique et epilepsie myoclonique benigne du nourrisson dans une meme famille: quelques reflexions sur la classification des epilepsies. Epilepsies 1996;8:307-15.**
Beaumanoir A, Blume W. The Lennox-Gastaut syndrome. In: Roger J, Bureau M, Dravet Ch, Genton P, Tassinari CA, Wolf P, editors. Epileptic syndromes in infancy, childhood and adolescence. 3rd ed. London: John Libbey Ltd, 2002:113-35.
Biondi R, Sofia V, Tarascone M, Leocata R. Epilessia mioclonica benigna dell'infanzia: contibuto clinico. Boll Lega Ut Epil 1991;74:93-4.
Colamaria V, Andrighetto G, Pinelli L, Olivieri A, Alfieri P, Dalla Bernardina B. Iperinsulinismo, ipoglicemia ed epilessia mioclonica benigna del lattante. Boll Lega It Epi 1987;58/59:231-3.
Commission on Classification and Terminology of the International League Against Epilepsy. Proposal for revised classification of epilepsies and epileptic syndromes. Epilepsia 1989;30:389-99.
Dalla Bernardina B, Colamaria V, Capovilla G, Bondavalli S. Nosological classification of epilepsies in the first three years of life. In: Nistico G, Di Perri R, Meinardi H, editors. Epilepsy: an update on research and therapy. New York: Alan Liss, 1983:165-83.
Delgado-Escueta AV, Greenberg D, Weissbecker K, et al. Gene mapping in the idiopathic generalized epilepsies: juvenile myoclonic epilepsy, childhood absence epilepsy, epilepsy with grand mal seizures, and early childhood myoclonic epilepsy. Epilepsia 1990;31(suppl 3):S19-29.
Doose H. Myoclonic astatic epilepsy of early childhood. In: Roger J, Bureau M, Dravet C, Dreifuss FE, Perret A, Wolf P, editors. Epileptic syndromes in infancy, childhood and adolescence, 2nd ed. London: John Libbey Eurotext Ltd., 1992:103-14.
Dravet C. Les epilepsies myocloniques benignes du nourrisson. Epilepsies 1990;2:95-101.
Dravet C, Bureau M. L'epilepsie myoclonique benigne du nourrisson. Rev Electroenceph Neurophysiol Clin 1981;11:438-44.
Dravet C, Bureau M, Giraud N, Roger J, Gobbi G, Dalla Bernardina B. Benign myoclonus of early infancy or benign non-epileptic spasms. Neuropediatrics 1986;17:33-8.
Dravet C, Bureau M. Benign myoclonic epilepsy in infancy. In: Roger J, Bureau M, Dravet Ch, Genton P, CA Tassinari, Wolf P, editors. Epileptic syndromes in infancy, childhood and adolescence. 3rd ed. London: John Libbey Ltd., 2002:69-79.**
Fusco L, Vigevano F. Ictal clinical and electroencephalographic findings of spasms in West syndrome. Epilepsia 1993;34:671-8.
Giovanardi Rossi P, Parmeggiani A, Posar A, Santi A, Santucci M. Benign myoclonic epilepsy: long-term follow-up of 11 new cases. Brain Dev 1997;19:473-9**
Guerrini R, Dravet CH, Gobbi G, Ricci S, Dulac O. Idiopathic generalized epilepsies with myoclonus in infancy and childhood. In: Malafosse A, Genton P, Hirsch E, Marescaux C, Broglin D, Bernasconi R, editors. Idiopathic generalized epilepsies: clinical, experimental, and genetic aspects. London: John Libbey Eurotext Ltd., 1994:267-80. **
Lin YP, Itomi K, Takada H, et al. Benign myoclonic epilepsy in infants: video-EEG features and long-term follow-up. Neuropediatrics 1998;29:268-71.
Loiseau P, Duche B, Loiseau J. Classification of epilepsies and epileptic syndromes in two different samples of patients. Epilepsia 1991;32:303-9.
Lombroso CT, Fejerman N. Benign myoclonus of early infancy. Ann Neurol 1977;1:138-43.
Pachatz C, Fusco L, Vigevano F. Benign myoclonus of early infancy. Epil Disord 1999;1:57-61.
Ricci S, Cusmai R, Fusco L, Cilio R, Vigevano F. Reflex myoclonic epilepsy of the first year of life. Epilepsia 1995;35:47.**
Todt H, Muller D. The therapy of benign myoclonic epilepsy in infants. In: Degen R, Dreifuss FE, editors. Epilepsy research. Suppl 6. The benign localized and generalized epilepsies in early childhood. Amsterdam: Elsevier, 1992:137-9.
Vigevano F, Cusmai R, Ricci S, Watanabe K. Benign epilepsies of infancy. In: Engel Jr J, Pedley TA, editors. Epilepsy: a comprehensive textbook. Philadelphia: Lippincott-Raven Publishers, 1997:2267-76.
Source - ILAE, Benign Myoclonic Epilepsy in Infancy
ILAE
Symptoms of myoclonic epilepsy
The clinical picture of myoclonic epilepsy depends on the type of disease. Among the main pathological symptoms are:
- Cramps. The disease is characterized by myoclonic seizures. They are not accompanied by severe pain. Cramps most often affect the limbs, less often the face and torso. On average, the duration of a seizure is 10-20 minutes. Consciousness is preserved.
- Loss of consciousness. Occurs extremely rarely. Characteristic of the youthful form.
- Tonic-clonic seizures. This form of seizure is accompanied by loss of consciousness and painful muscle contraction.
- Oligophrenia. Mental retardation occurs in various forms. Most often this is a disorder of creative thinking and intelligence.
- Mental disorders. Expressed by hallucinations, neuroses and borderline states.
Determining clinical symptoms is necessary to select the correct therapy. Doctors at the Yusupov Hospital develop an individual treatment plan for each patient.
Diagnosis of myoclonic epilepsy
Myoclonic epilepsy requires a comprehensive diagnosis. The examination includes:
- Collection of complaints and medical history. During the initial examination, the neurologist questions the patient about existing complaints, the time of their appearance, and the severity of symptoms. In addition, the duration of the attacks and the condition after them are specified.
- Neurological examination. The doctor evaluates reflexes and conducts tests to assess the state of nervous activity.
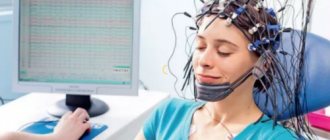
- EEG. Thanks to the study, it is possible to establish the pathological activity of various brain structures. The lesion site is determined in this way.
- MRI. To enhance the effect, the examination is carried out using contrast. Myoclonic epilepsy is not characterized by the presence of structural changes in the brain. However, there are exceptions.
The Yusupov Hospital uses modern medical equipment to diagnose myoclonic epilepsy. It allows you to quickly and effectively determine the presence of the disease. Based on the data obtained, correct therapy is prescribed.
Causes of the disease
Idiopathic epilepsy The only symptom of the disease is seizures. There are no other signs of neurological impairment. Idiopathic epilepsy is not associated with brain damage and is usually hereditary. Pathology can manifest itself and recede at a certain age. The International League Against Epilepsy calls this group of diseases “genetic.”
Symptomatic epilepsies The occurrence of seizures is caused by the presence of identifiable brain damage. Symptomatic forms are also called structural or metabolic.
Cryptogenic epilepsies The etiology has not been precisely established, so the pathologies cannot be unambiguously classified as symptomatic. Cryptogenic forms are called epilepsies of “unknown cause.”
Types of myoclonic epilepsy
Myoclonic epilepsy is divided into several types. Let's look at the main ones.
Infantile myoclonic epilepsy
Diagnosed in 30-40% of cases. Characterized by symptoms such as hyperkinesis or tics. A mild degree of development of the disease can go unnoticed for many years. Intellectual development does not suffer against the background of infantile myoclonic epilepsy. The disease can appear in a child from 2 months to 3 years.
Dravet syndrome
Identified in the first year of life. Symptomatically reminiscent of infantile myoclonic epilepsy. Dravet syndrome causes severe mental disorders. They manifest themselves as oligophrenia, mental retardation. Without correct treatment, the number of attacks increases to several times a week.
Unferricht-Lundborg disease
It is considered a genetic disease. Characterized by severe neurological symptoms. Unferricht-Lundborg disease is also accompanied by mental disorders. The first attack most often occurs during puberty. The disease is diagnosed in 10-20% of cases.
MERRF epilepsy
More often diagnosed in adults. Myoclonic epilepsy of adolescence is characterized by tonic-clonic paroxysms. The disease is not accompanied by neurological disorders or mental disorders. Consciousness remains clear during the attack.
Absence seizures are a type of epileptic seizure. Considered one of the symptoms of myoclonic epilepsy.
Depending on the progression of epilepsy, the following forms of the disease are distinguished:
- Progressive. The clinical picture of epilepsy is gradually increasing. As the disease progresses, the risk of death increases. In some cases, the disease is difficult to treat.
- Stable. Symptoms remain at approximately the same level.
- Remitting. Signs of epilepsy may develop slowly and then subside over a long period of time. Over time, complete disappearance of pathological signs is possible.
Case history and nomenclature
Benign myoclonic epilepsy syndrome in children (BMSES) was not clearly defined until it was first described in 7 young children in 1981 (Drave and Bior, 1981).
In this article, SDMED was defined as the occurrence of myoclonic seizures (MS) without seizures other than simple febrile seizures (FS) in the first three years of life in healthy children. These MPs were easily treatable and became quite rare during subsequent years of childhood. Psychomotor development remained normal, and no severe psychological consequences were observed. Since then, many other cases have been described in the literature. SDMED is included in the 1989 International Classification of Generalized Idiopathic Epilepsies (Commission, 1989). Some authors have described reflex MP in patients caused by noise or conversation, and proposed to distinguish between two separate nosological forms, and the 2nd was called reflex myoclonic epilepsy in children (Ricci et al., 1985). We think that such a division is inappropriate and will describe all cases as SDMED. To our knowledge, 98 cases have been described in the literature to date, of which 88 fit the classic description of SDMED and 10 were defined as “reflex SDMED.”
There are now 5 new patients under our supervision, four of whom have spontaneous and reflex seizures, which brings the total number of patients to 103.
The first description of SDMED indicated that the onset of the disease was before 3 years of age, while subsequent reports suggested a later onset in some patients, up to 4 years 8 months (Giovanardi-Rossi et al., 1997). This means that epilepsy of the same type can appear at different ages (Guerini et al., 1997).
Treatment of myoclonic epilepsy
Medications are used to treat myoclonic epilepsy. Doctors at the Yusupov Hospital use the following groups of drugs:
- antiepileptics;
- barbiturates;
- tranquilizers;
- nootropics.
An individual treatment plan is developed for each patient. It takes into account the form of myoclonic epilepsy, stage of development, age of the patient and the presence of concomitant diseases. This approach allows you to quickly and effectively select therapy. Experienced neurologists, epileptologists, and psychiatrists conduct consultations at the Yusupov Hospital. Rehabilitation is carried out by qualified massage therapists and exercise therapy instructors. In order to make an appointment, you need to call or leave a request on the official website of the hospital.
Treatment
VPA in monotherapy is the drug of choice, its use should be started as early as possible. It is preferable to use a solution rather than a syrup, as it is better tolerated by the child. It is necessary to strictly monitor the concentration of the drug in plasma.
A daily dose of 30 mg/kg 3 times a day is usually sufficient, but a higher dose may be required in some patients. VPA is also effective for possible febrile seizures. If VPA is ineffective, it can be used in combination with a benzoadiazepine (CLB or NTZ) or ETS and reconsider the diagnosis. Treatment should be continued for 3-4 years after the onset of the disease if it is well tolerated. In cases of purely reflex symptoms, drug therapy may not be used. If it has already started, it can be stopped abruptly, but if there is no hypersensitivity. If PEP occurs in a teenager, short-term treatment can be given at that age. Conservative treatment should be combined with psychological assistance to the family.