Niemann-Pick disease is a hereditary disease that is characterized by an increase in size and dysfunction of the liver and spleen, and in some cases, the development of mental disorders - epilepsy, dementia, developmental delays in children. The cause of these phenomena is the accumulation of fats in tissues with the gradual transformation of cells into a storage of sphingomyelin and cholesterol.
Scientists distinguish 4 types of Niemann-Pick disease, which are designated by the first letters of the Latin alphabet - A, B, C and D. Each of them is caused by various defects in the 11, 14 or 18 pairs of chromosomes. Type A is considered the most dangerous and malignant. It is diagnosed in the first months of life and without effective treatment, babies rarely manage to live more than two years.
Symptoms
The clinical picture of Niemann-Pick disease can be different, which led to its division into types. In more than 70% of cases, the pathology occurs as type B. In this case, the first symptom in infants is jaundice in the first months of life. By 2-3 years, a slight increase in the size of the liver and spleen and digestive disorders are recorded. At 6-8 years old, children often complain of abdominal pain, nausea and vomiting. Disruption of the spleen quickly affects the formation of platelets - in children with Niemann-Pick syndrome, blood clotting is impaired.
In the absence of effective treatment methods, fatty compounds begin to be deposited in the lungs. By adolescence, the patient’s chart shows frequent notes about bronchitis and pneumonia, and there are frequent cases of ascites - a sharp enlargement of the abdomen associated with disruption of the portal vein.
In type C, Niemann-Pick disease is also accompanied by disorders of the nervous system: children are developmentally delayed and often suffer from epileptic seizures and dementia. Symptoms of cirrhosis of the liver and obesity of the spleen progress so quickly that by the age of five a complex operation to remove their lobes may be required. Every year, treatment of Niemann-Pick disease abroad becomes more of a vital necessity, regardless of the cost and the need for flights.
Difficult diagnosis. Niemann–Pick disease, type C
L.S. NAMAZOVA-BARANOVA
1, 2, 3,
A.K.
GEVORGYAN 1, 2,, N.D.
VASHAKMADZE 1, L.S.
VYSOTSKAYA 1,
A.M.
MAMEDYAROV 1, MD,
T.V.
MARGIEVA 1, 2 1
Scientific Center for Children's Health of the Russian Academy of Medical Sciences, Moscow
2
First Moscow State Medical University named after.
THEM. Sechenov 3
Russian Scientific Research Medical University named after. N.I. Pirogova, Moscow In the conditions of modern diagnostic capabilities and the increasing level of knowledge of doctors, diseases that were previously considered extremely rare are increasingly being identified.
Along with the achievements of the pharmacological industry, timely diagnosis and the prescription of adequate therapy make it possible in many cases to save the life of a child and slow down the progression of the disease. The article is devoted to a rare, genetically determined pathology from the group of lysosomal diseases, inherited autosomal recessively - Niemann-Pick disease, type C. Variants of the clinical course and diagnostic methods are presented in detail. Storage diseases are a group of rare genetically determined diseases caused by impaired synthesis of lysosomal enzymes that control the intracellular breakdown of macromolecules (glycosaminoglycans, glycolipids, glycoproteins) and their intralysosomal accumulation, which leads to progressive dysfunction of damaged organs. The liver, spleen, central nervous system, and skeletal system are most often involved in the pathological process [1–4]. As a result of gene mutation, the activity of one or another enzyme decreases (up to 10–20% of normal), which is typical for mucopolysaccharidosis and glycogenosis. In rare cases, the so-called sorting of molecules inside cells. The latter is characteristic of Niemann-Pick disease, type C (NP-C), which is a sphingomyelin lipidosis that develops as a result of hereditary deficiency of proteins involved in intracellular lipid transport and a secondary decrease in sphingomyelinase activity (up to 20% of normal). The disease is inherited in an autosomal recessive manner. The NPC1 and NPC2 genes are localized in the 18q11-q12 and 14q24.3 loci. The disease is caused by a mutation in the NPC1 (95%) or NPC2 (4%) gene; in 1% of cases, the molecular genetic defect cannot be identified [1–9]. Classification
Depending on the molecular genetic defect, two types of the disease are distinguished: Niemann-Pick disease, type C, type 1 and Niemann-Pick disease, type C, type 2. Initially, Niemann–Pick disease, type D, detected in Nova Scotia isolates, was identified as a separate form, but later it was shown that mutations of the NPC1 gene are also the cause of this form of the disease [2, 5, 10]. Epidemiology
NP-C is one of the rare hereditary metabolic diseases. The disease is widespread. The incidence of the disease is 1: 120,000–1: 150,000 live newborns. A high frequency of type 1 NP-C disease was noted among some genetic isolates: the French colony of Acadia (Nova Scotia), Bedouin groups in Israel, Spanish settlements in Colorado and New Mexico (USA), which is associated with the founder effect [11, 12 ]. Clinical forms of Niemann–Pick disease, type C
The disease can manifest itself at any period of life - from prenatal to old age, but more often signs of NP-C appear in early childhood and quickly progress, leading to dysfunction of damaged organs. The course of the disease and the patient’s life expectancy largely depend on the age at which the onset occurred. The following forms of NP-C are distinguished: neonatal, infantile, late infantile, adolescent and adult [2, 11–13].
The neonatal form (onset before 3 months) is characterized by the development of intrauterine fetal hydrops. After birth, muscle hypotonia and delayed psychomotor development predominate, which progress and lead to intellectual impairment. In approximately half of the cases, intrahepatic cholestasis, jaundice and hepatosplenomegaly are noted, which may completely disappear with age. A fulminant course with the development of a severe form of cholestatic jaundice and an unfavorable outcome in the first half of life from liver failure is rarely observed. In a small number of cases, respiratory failure develops [9, 10, 13].
In the infant form (3 months - 2 years), the main clinical symptoms of NP-C (hepatosplenomegaly, delayed psychomotor development, muscle hypotonia) appear in the first year of life. Most patients never acquire independent walking skills. Intellectual development is usually not affected. As the disease progresses, the pyramidal system is involved in the pathological process, and a decline in cognitive functions gradually develops [11].
NP-C in 60–70% of cases usually manifests itself in late infancy and early childhood (late infantile form - 2–6 years). The first clinical symptoms of this form are cerebellar disorders (unsteadiness when walking, dysarthria, dysmetria), which usually appear at the age of 3–5 years. In 90% of cases, hepatosplenomegaly of varying severity is observed. As the disease progresses (including in the juvenile form), almost all patients develop vertical supranuclear ophthalmoparesis [14]. At the initial stages, the vertical movement of the eyeballs slows down, gradually progressing to a complete limitation of vertical and sometimes horizontal gaze. There is often a delay in speech development. Subsequently, previously acquired motor skills are gradually lost, intellectual impairments appear, dysphagia and dysarthria develop, and, less commonly, demyelinating peripheral polyneuropathy [14, 15]. An unfavorable outcome in the late infantile form usually occurs in the first decade of life [2, 13].
In the juvenile form of NP-C disease, the first symptoms usually appear between the ages of 6 and 15 years in the form of impaired learning of school material, writing, decreased attention, memory, and hyperactive behavior, which leads to an incorrect diagnosis. In some cases, the disease manifests itself with psychiatric disorders, such as behavioral disorders, schizophrenia-like syndrome, depression [2, 11]. In 20% of cases, gelastic cataplexy is observed: a short-term paroxysmal loss of muscle tone, leading to the patient falling without loss of consciousness, more often occurring against the background of strong emotional reactions (for example, during laughter) [16, 17]. Over time, cerebellar disorders progress, dysphagia, dysarthria appear, and intellectual development worsens. Frequent symptoms of NP-C are extrapyramidal disorders in the form of various dystonic hyperkinesis. In half of the cases, focal and/or generalized epileptic seizures develop, which are difficult to respond to adequate antiepileptic therapy. In the later stages of NP-C, pyramidal disorders increase in the form of increased muscle tone, revitalization of tendon reflexes, the appearance of pathological reflexes, and bulbar-pseudobulbar syndrome; Dementia, decerebrate or decortication rigidity develops. Hepatosplenomegaly is not usually observed. An unfavorable outcome most often develops at the end of the second – beginning of the third decade of life, usually from intercurrent infections [16–18].
Adult form. Symptoms of NP-C disease appear in the second or third decade of life, but can also occur after the age of 50 years [18]. The disease is characterized by a slowly progressive course. Cerebellar disorders (ataxia, dysarthria, dysmetria), muscular dystonia, and intellectual disorders of varying severity gradually develop. Often in adults, NP-C disease manifests itself with the development of bipolar disorders [16–18]. In this form, epileptic seizures rarely develop; an enlarged spleen and vertical supranuclear ophthalmoparesis are not typical.
Diagnostics
Currently, the main methods for diagnosing NP-C are biochemical tests that reveal disturbances in intracellular transport and cholesterol homeostasis. The presence of mutations in the NPC1 or NPC2 genes in combination with characteristic clinical manifestations confirms the diagnosis. Additional studies are needed to study new mutations [1, 2, 5, 6, 9]. To diagnose the disease using biochemical methods, the presence of living cells (usually a culture of skin fibroblasts) is required.
The filipin staining test is considered the most sensitive and specific method. Fibroblasts are cultured in a medium rich in low-density lipoproteins, after which the cells are fixed and stained with filipin. In 85% of cases (“classical biochemical phenotype”), fluorescence microscopy reveals numerous fluorescent (cholesterol-filled) perinuclear vesicles in the studied cells. Less pronounced accumulation of cholesterol is observed in 15% of cases (“variant biochemical phenotype”). Interpretation of microscopy results in such cases is often difficult, and the results can be both false-positive and false-negative [19, 20].
The second most informative test is measuring the rate of cholesterol ester formation induced by low-density lipoproteins in fibroblast culture [19, 20]. In cell lines from patients with the “classical biochemical phenotype,” the rate of cholesterol esterification is close to or zero; with a “variant biochemical phenotype,” a slight disturbance in the esterification of cholesterol is observed. Accordingly, the test results may be ambiguous. In such cases, genetic mutation analysis is necessary to confirm the diagnosis. Considering the complexity, high cost and labor-intensiveness of biochemical research, when a test with filipin is positive, mutation analysis is often performed immediately [19, 20].
Biochemical tests are not informative enough in the case of heterozygous carriers of NP-C disease: the results of a test with filipin may be normal; mild abnormalities similar to those in “variant” cell lines are observed [20, 21].
Examination of bone marrow smears stained with filipin (as well as Giemsa) reveals foam cells filled with cholesterol and is considered a rapid method for screening NP-C, but does not make it possible to establish a final diagnosis [20, 21].
For prenatal diagnosis of NP-C, the molecular genetic method should be used [5].
Histological methods involve examining tissue biopsies using light and electron microscopy. Detect characteristic (but nonspecific) foam cells and/or blue histiocytes in the bone marrow, spleen, liver, lungs and lymph nodes (by light microscopy), in some cases revealing hidden lesions in the skin, skeletal muscles and eyes [16, 22, 23] . The absence of changes in light microscopy does not exclude the diagnosis of NP-C disease (for example, manifested by cholestatic liver damage in newborns).
The detection of polymorphic cytoplasmic inclusions during electron microscopy of liver or skin biopsies is pathognomonic for NP-C; however, they are not always detected [5, 16, 22].
Genetic testing is the gold standard for final verification of the diagnosis, as well as for prenatal diagnosis [5, 16]. Molecular genetic research of the NPC1 and NPC2 genes is carried out in specialized laboratories. In some cases, to identify an NPC1 mutation, it is necessary to perform a combined gDNA and cDNA study [5, 6, 16, 23]. Carrier identification and prenatal diagnosis
If the diagnosis of NP-C disease is confirmed, it is necessary to carry out genetic counseling of the patient: to clarify the nature of the disease, the type of inheritance, and provide recommendations for family planning (with prenatal diagnosis). It is recommended to test DNA from both parents.
Prenatal diagnosis can be established based on the study of chorionic villi at 10–12 weeks of pregnancy. Molecular genetic analysis is considered preferable, for which (unlike biochemical prenatal diagnosis) does not require cell culture [5].
The heterogeneity and variability of the clinical manifestations of NP-S depending on the age of manifestation determine the underdiagnosis of the disease. This is obvious from the example of our country: at the beginning of 2010 in the Russian Federation, only 2 patients were diagnosed with Niemann-Pick disease, type C [24], but also subsequently, despite numerous publications on this topic in medical periodicals , conducting scientific and practical seminars, only 7 cases of this pathology were registered [2, 10, 24, 25]. Considering the prevalence of the disease, it is obvious that there should be more patients with NP-C disease in the country. This means that in the absence of specific therapy they are at risk of rapid progression of the disease, disability and, worst of all, an earlier onset of an unfavorable outcome. While early administration of specific therapy allows to slow down the further development of the disease. Currently, the best evidence base for efficacy and safety is the drug miglustat (Zaveska, Actelion Pharmaceuticals Ltd, Switzerland). This is the only drug approved for use in NP-C. Miglustat is a small molecule (iminosugar) that competitively inhibits the enzyme glucosylceramide synthetase, which catalyzes the first step in glycosphingolipid synthesis [26–28], thereby suppressing the accumulation of the neurotoxic gangliosides GM2 and GM3, lactosylceramide and glucosylceramide. In the European Union (EU), the USA and some other countries, miglustat is used to treat patients with mild or moderate forms of type 1 Gaucher disease who are not eligible for enzyme replacement therapy [26–28]. Miglustat was registered for the treatment of NP-C disease in 2006 in the EU countries, and in 2008 in the USA. In January 2009, the EU Commission expanded the indications for the use of miglustat: the treatment of progressive neurological manifestations of NP-C in adults and children [2].
Since the time of initiation of specific and adequate symptomatic therapy is crucial for the prognosis of NP-C disease, it is important to identify the disease as early as possible - to establish a diagnosis. This problem prompted a group of researchers led by Professor FA Wijburg to create an algorithm for diagnosing NP-C disease [29].
Scientists have set themselves the following goals:
1) develop a prognostic tool “index of suspicion of NP-C disease” and validate the specificity and sensitivity of indicators based on the analysis of patient records; 2) develop a scoring scale for assessing symptoms and their combinations to assess the likelihood of diagnosing NP-C disease; 3) to develop a simple tool that could allow doctors of various specialties who are not familiar with the problem of NP-C disease: - understand the main symptoms and manifestations, - suggest the patient has NP-C disease as a possible diagnosis.
In this study, all symptoms of NP-C disease were divided into three main categories: visceral, neurological and psychiatric. A retrospective analysis of data from 216 patients referred for examination with suspected NP-C disease to 7 centers in Europe and Australia allowed us to assess the diagnostic importance of each individual symptom and their totality. Based on the results of statistical analysis, symptoms within each category were divided into 5 groups according to the degree of likelihood of NP-C disease: from the most likely (40 points/point), such as vertical supranuclear gaze palsy and gelastic cataplexy, to unlikely or additional (1 point/point). point), such as hypotension, seizures and myoclonus. For example, ataxia occurs in approximately 80% of patients with Niemann-Pick disease, type C [1], as a symptom in this algorithm is assessed at 10 points (average probability index). A similar symptom can occur in many other hereditary diseases, that is, its specificity is low. At the same time, unexplained isolated splenomegaly, which occurs in approximately 50% of cases of NP-C disease [20], is a highly specific symptom of this pathology (20 points).
In the “visceral symptoms” section, medical history data are also taken into account. Despite the possibility of complete resolution during the first year of life of prolonged jaundice and hepatosplenomegaly (if present in patients in the neonatal period), these symptoms are important for making a diagnosis.
During the development of the diagnostic algorithm, the relationship between symptoms was assessed. The presence of symptoms from the categories “neurological” and “visceral” at the same time in a patient increases the prognostic score of NP-C by 40, as does the combination of visceral and psychiatric symptoms. The combination of neurological and psychiatric symptoms is scored 20 points. Family risk is additionally assessed: 40 points - if you have siblings or parents with Niemann-Pick disease, type C, 20 points - if you have cousins/sisters with this pathology.
The total assessment or prognostic score allows the doctor to develop tactics for further management of the patient:
<40 points—the diagnosis of NP-C disease is unlikely; 40–69 points—further observation and consultation at a specialized center for differential diagnosis are necessary; ≥70 points - a high probability of NP-C disease; it is necessary to urgently refer the patient to a specialized center for diagnosis.
Bibliography
1. Vanier MT, Millat G. Niemann–Pick disease type C. Clin. Genet. 2003; 64:269–281. 2. Wraith J, Baumgartner M, Bembi B. Recommendations for the diagnosis and treatment of Niemann-Pick disease type C. Pediatric pharmacology. 2010; 7 (1): 16–24. 3. URL: https://www.esgld.org/ (European Study Group on Lysosomal Diseases - ESGLD). 4. URL: https://www.hgmd.cf.ac.uk HGMD (Human Gene Mutation Database, Cardiff). 5. Vanier MT. Prenatal diagnosis of Niemann–Pick diseases types A, B and C. Prenat. Diagn. 2002; 22: 630–632. 6. Millat G, Bailo N, Molinero S et al. Niemann–Pick C disease: use of denaturing high performance liquid chromatography for the detection of NPC1 and NPC2 genetic variations and impact on management of patients and families. Mol. Genet. Metab. 2005; 86:220–232. 7. Subramanian K, Balch WE. NPC1/NPC2 function as a tag team duo to mobilize cholesterol. Proc. Natl. Acad. Sci. 2008; 105:15223–15224. 8. Sleat DE, Wiseman JA, El-Banna M et al. Genetic evidence for nonredundant functional cooperation between NPC1 and NPC2 in lipid transport. Proc. Natl. Acad. Sci. 2004; 101:5886–5891. 9. Fernandez-Valero E, Ballart A, Itturiaga C et al. Identification of 25 new mutations in 40 unrelated Spanish Niemann–Pick type C patients: genotype-phenotype correlations. Clin. Genet. 2005; 68:245–254. 10. Mikhailova S.V., Zakharova E.Yu., Bukina T.M. and others. Niemann-Pick disease type C: Clinical examples. Pediatric pharmacology. 2010; 7 (5): 48–53. 11. Scriver CR et al. The Metabolic and molecular bases of inherited diseases (McGraw-Hill). New York, 2005. 12. Imrie J, Dasgupta S, Besley GT et al. The natural history of Niemann–Pick disease type C in the UK. J. Inherit. Metab. Dis. 2007; 30:51–59. 13. Kelly DA, Portmann B, Mowat AP et al. Niemann–Pick disease type C: diagnosis and outcome in children, with particular reference to liver disease. J. Pediatr. 1993; 123:242–247. 14. Solomon D, Winkelman AC, Zee DS et al. Niemann–Pick type C disease in two affected sisters: ocular motor recordings and brainstem neuropathology. Ann. NY Acad. Sci. 2005; 1039:436–445. 15. Zafeiriou DI, Triantafyllou P, Gombakis NP et al. Niemann–Pick type C disease associated with peripheral neuropathy. Pediatr. Neurol. 2003; 29: 242–244. 16. Patterson MC, Vanier MT, Suzuki K et al. The Metabolic and molecular bases of inherited disease (McGraw-Hill). New York, 2001. 3611–3633. 17. Sevin M, Lesca G, Baumann N et al. The adult form of Niemann–Pick disease type C. Brain. 2007; 130: 120–133. 18. Bauer P, Boettcher T, Meyer WP et al. Niemann–Pick type C disease (NP-C) is a significant diagnosis in juvenile and adult-onset psychiatric disorders. Annual Meeting of the American Society of Human Genetics (ASH-G), Philadelphia, USA, 2008. 19. Argoff CE, Comly ME, Blanchette-Mackie J et al. Type C Niemann–Pick disease: cellular uncoupling of cholesterol homeostasis is linked to the severity of disruption in the intracellular transport of exogenously derived cholesterol. Biochim. Biophys. Acta. 1991; 1096:319–327. 20. Vanier MT, Rodriguez-Lafrasse C, Rousson R et al. Type C Niemann–Pick disease: spectrum of phenotypic variation in disruption of intracellular LDL-derived cholesterol processing. Biochim. Biophys. Acta. 1991; 1096:328–337. 21. Strauss JF, Liu P, Christenson LK et al. Sterols and intracellular vesicular trafficking: lessons from the study of NPC1. Steroids. 2002; 67:947–51. 22. Vanier MT, Millat G. Structure and function of the NPC2 protein. Biochim. Biophys. Acta. 2004; 1685: 14–21. 23. Paciorkowski AR, Westwell M, Ounpuu S et al. Motion analysis of a child with Niemann–Pick disease type C treated with miglustat. Mov. Discord. 2008; 23: 124–128. 24. Rudenskaya G.E., Zakharova E.Yu., Bukina T.M. and others. Niemann-Pick disease, type C (juvenile dystonic lipidosis). Journal neurol. and a psychiatrist. them. S.S. Korsakov. 2008; 5:76–79. 25. Niemann's disease - Pick type With the eyes of a mother, nurse, doctor. Pediatric pharmacology. 2010; 7 (2): 149–150. 26. Butters TD, Dwek RA, Platt FM. Inhibition of glycosphingolipid biosynthesis: application to lysosomal storage disorders. Chem. Rev. 2000; 100:4683–4696. 27. Wraith JE, Vecchio D, Jacklin E et al. Disease stability in patients with Niemann–Pick disease type C treated with miglustat, Lysosomal diseases network WORLD symposium 2009. San Diego, California, USA, 2009. 28. Wraith JE, Pineda M, Sedel F et al. Miglustat in patients with Niemann–Pick Type C disease (NP-C): a multicentre retrospective survey. Annual Meeting of the American Society of Human Genetics, Philadelphia, USA, 2008. 29. Wijburg FA et al. NP-C disease, an autosomal recessive neurovisceral cellular lipidosis, is characterized by the inappropriate movement and partitioning of LDL-derived cholesterol. Cell. Res. 2005; 255:56–66.
Diagnosis of the disease
As a rule, typical symptoms of the disease can be noticed already in the first months of the baby’s life. Enlarged liver and spleen, indifference to the environment and lethargy are the first signs of pathology, when detected, the child is sent for tests to a genetics center. Nowadays a whole range of techniques is used that allows not only to establish a diagnosis, but also to determine the type of disease and make predictions.
- In skin cell culture, defects in 11, 14 or 18 pairs of genes are detected;
- analyze the activity of sphingomyelinase in a culture of leukocytes;
- The most accurate results are obtained from a bone marrow biopsy, which reveals characteristic “foam” cells.
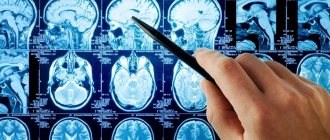
Treatment of Niemann-Pick disease abroad does not begin without additional diagnostics necessary to determine the extent of anatomical changes in organs and tissues. A computed tomography scan of the brain, an ultrasound scan of the abdominal cavity, and psychiatric tests are required.
Etiology of the development of Niemann-Pick pathology
The main reason for the development of Niemann-Pick pathology is a defect in chromosomes:
- On chromosome No. 11 (pathology type A);
- On chromosomes No. 14 and No. 18 (pathology type B).
When there is a violation or defect in the chromosomes, there is a decrease in the activity of sphingomyelinase molecules, which breaks down sphingomyelin fat molecules.
With such a disturbance in activity, sphingomyelin fat molecules accumulate together with cholesterol molecules in macrophages, which leads to disruption of lipid metabolism, as well as to the metabolism of the entire body.
Provoking factors that can aggravate the progression of Niemann-Pick disease:
- Poor nutrition with a predominance of animal fats in the diet;
- Alcohol abuse;
- Physical inactivity and complete absence of stress on the body;
- Overweight - obesity;
- Constant overstrain of the nervous system;
- Frequent stressful situations;
- Chronic pathologies in the body.
If several genes are mutated simultaneously, then Niemann-Pick disease occurs in a complicated form.
When there is a violation or defect in the chromosomes, the activity of sphingomyelinase molecules decreases
Treatment of pathology in Germany
For many years, only a number of medications were used to treat Niemann-Pick disease to alleviate the symptoms of the disease. In chronic cases of types B and C, this approach made it possible to significantly improve the patient’s quality of life and prolong his life.
Today, treatment of Niemann-Pick disease in Germany is based on the latest advances in medicine and genetics. Drugs that block the synthesis of sphingomyelin, the accumulation of which is the cause of enlargement of internal organs and mental disorders, are widely used. Their use is indicated for patients throughout their lives.
High results are achieved by bone marrow transplantation, in which stem cells with a defective genotype are replaced with healthy ones.
The timing of a transplant is determined not so much by the price as by the time it takes to find a suitable donor. Having healthy relatives with a suitable set of immunoglobulins simplifies the task. If the organs are significantly enlarged, surgery is performed to remove the spleen and resection of the liver.
Prices for Niemann-Pick disease treatment in Germany vary depending on the number of procedures and surgeries required. A benign course of type B extremely rarely requires a surgical solution, while type A requires urgent qualified intervention.
Hereditary neuromuscular diseases
Hereditary neuromuscular diseases are represented by the most extensive group of pathologies.
Group 1 of neuromuscular dystrophy includes spinal amyotrophies. Common forms of spinal amyotrophy include:
- progressive spinal Aran-Duchenne amyotrophy, observed in adults;
- Kugelberg-Welander juvenile amyotrophy;
- childhood form of Werdnig-Hoffmann amyotrophy.
The second group of neuromuscular dystrophy is represented by neural amyotrophies. These diseases can be either familial or sporadic. This group of diseases usually manifests itself in childhood or adolescence. This group of neuromuscular dystrophies includes:
- Roussy-Lévy syndrome;
- neural form of amyotrophy Charcot-Marie-Tooth;
- Refsum's disease;
- hypertrophic interstitial neuropathy Dejerine-Sotta.
A large group of neuromuscular diseases is represented by primary progressive muscular dystrophies. Manifestations of primary progressive muscular dystrophies can be varied. Prominent representatives of this group of diseases are:
- pseudohypertrophic childhood dystrophy Duchenne;
- favorable current pseudohypertrophic dystrophy of Becker-Keener;
- juvenile and end-lumbar Erb's dystrophy;
- distal muscular dystrophy;
- oculopharyngeal and ocular forms of dystrophy;
- rigid spine syndrome;
- non-progressive muscular dystrophy.
- hereditary paramyotonia Eulenburg;
- Thomsen's myotonia;
- neuromyotonia;
- dystrophic myotonia.
Group 5 neuromuscular dystrophies includes myoplegic syndromes and paroxysmal myoplegia. The most common myoplegias include:
- normokalemic myoplegia;
- Hamstorp disease;
- hypokalemic paroxysmal myoplegia;
- secondary variants of paroxysmal myoplegia.
All these diseases have their own characteristics of course and development. Some of them allow patients to live a full life and not experience obvious discomfort, while others lead to significant disruption of the muscular system.
DMU: comprehensive treatment support in Germany
Severe syndromes and a threat to the life of a child with Niemann-Pick disease dictate their conditions to parents - treatment must be provided regardless of its price, with high quality and without delay. If at the same time you are stopped by a difficult choice and search for a clinic, lack of language knowledge, fear of flying, the need for accommodation in a foreign country and the difficulty of obtaining visas, our specialists are ready to help and take responsibility for all organizational issues. After all, Niemann-Pick disease in Germany is considered and treated comprehensively, which makes it possible to compensate for the pathology at any age.
Specialists from the DMU center in Russia will be by your side throughout the entire treatment cycle:
- we will select the best specialists in Germany for you, provide an estimate, treatment plan and schedule a visit;
- We will help you with obtaining a medical visa;
- we will book tickets and accompany you from departure to return home;
- There will be a professional translator next to you who will explain everything the doctor says.
You can be sure that the prices for each service are justified. Our coordinators carefully check the invoices issued and compare them with the price list recommended by the German Ministry of Health. Treatment abroad can be your ticket to a new life, and we are ready to support you in every action.
Classification
There are three clinical forms of this disease:
- type A is the classic infantile form. Symptoms appear already in the first year of life and are characterized by neurological disorders (convulsions, difficulty swallowing, lack of reaction). In most cases, children die before the age of 3;
- type B – visceral form. Symptoms may appear between the ages of 2 and 6 years. The liver and spleen are most affected. The risk of death is much lower, many patients live to an old age;
- type C - adolescent non-acute form. Symptoms appear at the age of 2–5 years, becoming more intense at 15–18 years. Internal organs are affected, including the brain. Most often, patients die at 15–18 years of age.
The visceral form of the disease is considered the most favorable, since death is diagnosed extremely rarely, and the clinical picture is less pronounced.
What are the forecasts?
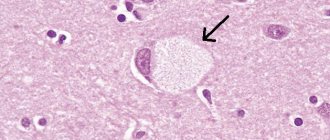
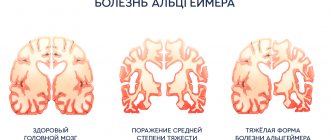
Unfortunately, the prognosis for patients with this diagnosis is disappointing. Pick's disease is an extremely serious problem, and to date there is no way to stop the destruction of neurons. However, the use of appropriate medications and high-quality rehabilitation help slow down the process, maintaining the patient’s quality of life.
But, according to statistics, within 5–10 years the disease leads to mental and moral decomposition of the individual - patients develop dementia, marasmus, cachexia, etc. Such patients should be under constant supervision, preferably in specialized clinics, where they will be constantly monitored by medical staff.
Is there an effective treatment?
What treatment does Pick's disease require? Treatment today boils down to attempts to stop further progression of the disease. First of all, patients need replacement therapy - patients are prescribed substances that cannot be synthesized in the body due to atrophy of nerve tissue (for example, acetylcholinesterase inhibitors). In some cases, additional intake of anti-inflammatory substances is necessary.
An extremely important part of therapy is the intake of neuroprotectors, which stimulate the vital activity of neurons. In the presence of mental disorders, appropriate medications are used to help reduce symptoms, for example, eliminate aggression. Sometimes it is necessary to take antidepressants.
Of course, an important part of treatment is psychological support. Patients are recommended to regularly attend special classes and trainings that help slow down the process of mental and emotional changes.
What symptoms accompany the further development of the disease?
As the disease progresses, symptoms become more severe. Often a person has speech disorders - the vocabulary becomes more meager and over time is reduced to a few phrases that the patient repeats constantly. Naturally, this also affects writing. Signs of Pick's disease include aphasia, alexia, agraphia, etc.
In addition, in some cases, the disease is accompanied by the appearance of hypokinesis, loss of muscle tone, etc. Quite often, patients with a similar diagnosis suffer from obesity and also refuse to observe personal hygiene rules.
In any case, at later stages the patient needs help - the person is no longer able to take care of himself.