Болезнь Ниманна-Пика – наследственное заболевание, которое характеризуется увеличением в размерах и нарушением функций печени и селезенки, а в ряде случаев и развитием психических расстройств – эпилепсии, слабоумия, отставания детей в развитии. Причиной этих явлений становится накопление в тканях жиров с постепенным преображением клеток в хранилище сфингомиелина и холестерина.
Ученые выделяют 4 типа болезни Ниманна-Пика, которые обозначаются первыми буквами латинского алфавита – A, B, C и D. Каждый из них обусловлен различными дефектами в 11, 14 или 18 паре хромосом. Наиболее опасным и злокачественным считается тип А. Его диагностируют в первые месяцы жизни и без эффективного лечения малышам редко удается прожить более двух лет.
Симптоматика
Клиническая картина болезни Ниманна-Пика может быть различной, что и обусловило деление ее на типы. Более чем в 70% случаев патология протекает по типу B. При этом первым симптомом у младенцев становится желтуха в первые месяцы жизни. К 2-3 годам регистрируют некоторое увеличение в размерах печени и селезенки, нарушение пищеварения. В 6-8 лет дети часто жалуются на боли в животе, тошноту и рвоту. Нарушение работы селезенки быстро сказывается на формировании тромбоцитов – у детей с синдромом Ниманна-Пика нарушается свертываемость крови.
В отсутствие эффективных методов лечения, жировые соединения начинают откладываться и в легких. К подростковому возрасту у больного в карте появляются частые отметки о бронхитах и пневмониях, нередки случаи асцита – резкого увеличения живота, связанного с нарушением работы воротной вены.
При типе С болезнь Ниманна-Пика сопровождается еще и расстройствами нервной системы: дети отстают в развитии, нередко страдают от эпилептических припадков и слабоумия. Симптомы цирроза печени и ожирения селезенки при этом прогрессируют настолько быстро, что уже к пяти годам может потребоваться сложная операция по удалению их долей. С каждым годом лечение болезни Ниманна-Пика за рубежом становится большей жизненной необходимостью, невзирая на цену и необходимость перелетов.
Трудный диагноз. Болезнь Ниманна – Пика, тип С
Л.С. НАМАЗОВА-БАРАНОВА
1, 2, 3,
А.К. ГЕВОРКЯН
1, 2,, Н.Д. ВАШАКМАДЗЕ 1, Л
.С. ВЫСОЦКАЯ
1,
А.М. МАМЕДЬЯРОВ
1, д.м.н.,
Т.В. МАРГИЕВА
1, 2 1
Научный центр здоровья детей РАМН, Москва
2
Первый Московский государственный медицинский университет им. И.М. Сеченова
3
Российский научный исследовательский медицинский университет им. Н.И. Пирогова, МоскваВ условиях современных диагностических возможностей и повышения уровня знаний врачей все чаще выявляют болезни, которые ранее считались крайне редкими. Наряду с достижениями фармакологической промышленности, своевременная диагностика и назначение адекватной терапии позволяют во многих случаях сохранить жизнь ребенку и замедлить прогрессирование болезни. Статья посвящена редкой, генетически обусловленной патологии из группы лизосомных болезней, наследуемой аутосомно-рецессивно, — болезни Ниманна – Пика, тип С. Подробно представлены варианты клинического течения и методы диагностики.
Болезни накопления — группа редких генетически детерминированных болезней, обусловленных нарушением синтеза лизосомных ферментов, контролирующих внутриклеточное расщепление макромолекул (гликозаминогликанов, гликолипидов, гликопротеинов) и внутрилизосомным их накоплением, что приводит к прогрессирующему нарушению функции поврежденных органов. Наиболее часто в патологический процесс вовлекаются печень, селезенка, центральная нервная система, костная система [1–4]. В результате мутации гена снижается активность того или иного фермента (до 10–20% нормы), что характерно для мукополисахаридозов и гликогенозов. В редких случаях нарушается т. н. сортировка молекул внутри клеток. Последнее свойственно болезни Ниманна – Пика, тип С (НП-С), которая представляет собой сфингомиелиновый липидоз, развивающийся в результате наследственной недостаточности белков, участвующих во внутриклеточном транспорте липидов, и вторичного снижения активности сфингомиелиназы (до 20% нормы). Заболевание наследуется по аутосомно-рецессивному типу. Гены NPC1 и NPC2 локализуются в локусах 18q11-q12 и 14q24.3. Болезнь обусловлена мутацией гена NPC1 (95%) или NPC2 (4%), в 1% случаев молекулярно-генетический дефект идентифицировать не удается [1–9]. Классификация
В зависимости от молекулярно-генетического дефекта выделяют два типа заболевания: болезнь Ниманна – Пика, тип С, 1-го типа и болезнь Ниманна – Пика, тип С, 2-го типа. Первоначально выделяли в отдельную форму болезнь Ниманна – Пика, тип D, выявляемую в изолятах Новой Шотландии, но в дальнейшем было показано, что причиной этой формы болезни также являются мутации гена NPC1 [2, 5, 10]. Эпидемиология
НП-С относится к числу редких наследственных болезней обмена веществ. Заболевание распространено повсеместно. Частота заболевания — 1 : 120 000–1 : 150 000 живых новорожденных. Высокая частота болезни НП-С 1-го типа отмечена среди некоторых генетических изолятов: французской колонии Акадия (Новая Шотландия), групп бедуинов в Израиле, испанских поселений в Колорадо и Нью-Мексико (США), что связано с эффектом основателя [11, 12]. Клинические формы болезни Ниманна – Пика, тип С
Болезнь может манифестировать в любом периоде жизни — от внутриутробного до пожилого возраста, но чаще признаки НП-С появляются в раннем детском возрасте и быстро прогрессируют, приводя к нарушению функций поврежденных органов. Течение болезни и продолжительность жизни пациента во многом зависят от возраста, в котором произошел дебют. Выделяют следующие формы НП-С: неонатальную, младенческую, позднюю младенческую, юношескую и взрослую [2, 11–13].
Неонатальная форма (дебют до 3 мес.) характеризуется развитием внутриутробной водянки плода. После рождения преобладает мышечная гипотония и задержка психомоторного развития, которые прогрессируют и приводят к нарушениям интеллекта. Приблизительно в половине случаев отмечаются внутрипеченочный холестаз, желтуха и гепатоспленомегалия, которые с возрастом могут полностью исчезнуть. Редко наблюдается молниеносное течение с развитием тяжелой формы холестатической желтухи и неблагоприятным исходом на первом полугодии жизни от печеночной недостаточности. В небольшом числе случаев развивается дыхательная недостаточность [9, 10, 13].
При младенческой форме (3 мес. – 2 года) основные клинические симптомы НП-С (гепатоспленомегалия, задержка психомоторного развития, мышечная гипотония) появляются на первом году жизни. Большинство пациентов никогда не приобретают навыки самостоятельной ходьбы. Интеллектуальное развитие обычно не страдает. По мере прогрессирования болезни происходит вовлечение в патологический процесс пирамидной системы, постепенно развивается снижение когнитивных функций [11].
НП-С в 60–70% случаев обычно манифестирует в позднем младенческом и раннем детском возрасте (поздняя младенческая форма — 2–6 лет). Первыми клиническими симптомами этой формы являются мозжечковые расстройства (шаткость при ходьбе, дизартрия, дисметрия), которые обычно проявляются в возрасте 3–5 лет. В 90% случаев наблюдается гепатоспленомегалия различной степени выраженности. По мере прогрессирования болезни (в т. ч. при юношеской форме) практически у всех пациентов развивается вертикальный супрануклеарный офтальмопарез [14]. На начальных этапах происходит замедление движения глазных яблок по вертикали, постепенно прогрессирующее до полного ограничения вертикального, а иногда и горизонтального взора. Нередко отмечается задержка речевого развития. В дальнейшем постепенно утрачиваются ранее приобретенные двигательные навыки, проявляются интеллектуальные нарушения, развивается дисфагия и дизартрия, реже демиелинизирующая периферическая полинейропатия [14, 15]. Неблагоприятный исход при поздней младенческой форме обычно наступает в первую декаду жизни [2, 13].
При юношеской форме болезни НП-С первые симптомы обычно появляются в возрасте от 6 до 15 лет в виде нарушения усвоения школьного материала, письма, снижения внимания, памяти, гиперактивного поведения, что приводит к установлению неправильного диагноза. В некоторых случаях болезнь манифестирует с психиатрических нарушений, таких как нарушение поведения, шизофреноподобный синдром, депрессия [2, 11]. В 20% случаев наблюдается геластическая катаплексия: кратковременная приступообразная утрата мышечного тонуса, приводящая к падению больного без потери сознания, чаще возникающая на фоне сильных эмоциональных реакций (например, во время смеха) [16, 17]. Со временем прогрессируют мозжечковые расстройства, появляются дисфагия, дизартрия и ухудшается интеллектуальное развитие. Частыми симптомами НП-С являются экстрапирамидные расстройства в виде различных дистонических гиперкинезов. В половине случаев развиваются фокальные и/или генерализованные эпилептические приступы, трудно поддающиеся адекватной антиэпилептической терапии. На поздних стадиях НП-С нарастают пирамидные нарушения в виде повышения мышечного тонуса, оживления сухожильных рефлексов, появления патологических рефлексов, бульбарно-псевдобульбарного синдрома; развивается деменция, децеребрационная или декортикационная ригидность. Гепатоспленомегалия обычно не наблюдается. Неблагоприятный исход чаще развивается в конце второго – начале третьего десятилетия жизни, обычно от интеркуррентных инфекций [16–18].
Взрослая форма. Симптомы болезни НП-С появляются на втором-третьем десятилетии жизни, но могут возникнуть и в возрасте старше 50 лет [18]. Характерно медленно прогрессирующее течение болезни. Постепенно развиваются мозжечковые расстройства (атаксия, дизартрия, дисметрия), мышечная дистония, различной степени выраженности интеллектуальные расстройства. Нередко у взрослых болезнь НП-С манифестирует с развития биполярных расстройств [16–18]. При этой форме эпилептические приступы развиваются редко, не характерно увеличение селезенки и вертикальный надъядерный офтальмопарез.
Диагностика
В настоящее время основными методами диагностики НП-С считают биохимические тесты, позволяющие выявить нарушение внутриклеточного транспорта и гомеостаза холестерина. Наличие мутаций NPC1- или NPC2- генов в сочетании с характерными клиническими проявлениями подтверждает диагноз. Для изучения новых мутаций необходимы дополнительные исследования [1, 2, 5, 6, 9]. Для диагностики болезни биохимическими методами требуется наличие живых клеток (обычно культуры фибробластов кожи).
Самым чувствительным и специфичным методом считают тест с окрашиванием филипином. Фибробласты культивируют в среде с большим содержанием липопротеидов низкой плотности, после чего клетки фиксируют и окрашивают филипином. В 85% случаев («классический биохимический фенотип») при флюоресцентной микроскопии в исследуемых клетках обнаруживают многочисленные флюоресцирующие (заполненные холестерином) перинуклеарные пузырьки. Менее выраженное накопление холестерина отмечается в 15% случаев («вариантный биохимический фенотип»). Интерпретировать результаты микроскопии в таких случаях часто трудно, а результаты могут быть как ложноположительными, так и ложноотрицательными [19, 20].
Второй по информативности тест — измерение скорости образования эфира холестерина, индуцированного липопротеинами низкой плотности, в культуре фибробластов [19, 20]. В клеточных линиях пациентов с «классическим биохимическим фенотипом» скорость этерификации холестерина близка или равна нулю; при «вариантном биохимическом фенотипе» наблюдается легкое нарушение этерификации холестерина. Соответственно, результаты теста могут оказаться неоднозначными. В таких случаях для подтверждения диагноза необходим анализ генетических мутаций. Учитывая сложность, высокую стоимость и трудоемкость биохимического исследования, при положительной пробе с филипином часто сразу проводят анализ мутаций [19, 20].
Биохимические тесты недостаточно информативны в случае гетерозиготных носителей болезни НП-С: результаты пробы с филипином при этом могут быть нормальными; наблюдаются легкие нарушения, сходные с таковыми в «вариантных» клеточных линиях [20, 21].
Исследование мазков костного мозга, окрашенных филипином (а также по Гимзе), позволяет выявить пенистые клетки, заполненные холестерином, считается экспресс-методом скрининга НП-С, однако не дает возможности установить окончательный диагноз [20, 21].
Для пренатальной диагностики НП-С следует использовать молекулярно-генетический метод [5].
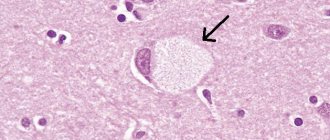
Гистологические методы предполагают исследование биоптатов тканей с помощью световой и электронной микроскопии. Обнаруживают характерные (но неспецифические) пенистые клетки и/или голубые гистиоциты в костном мозге, селезенке, печени, легких и лимфатических узлах (при световой микроскопии), в некоторых случаях выявляя скрытое поражение кожи, скелетных мышц и глаз [16, 22, 23]. Отсутствие изменений при световой микроскопии не исключает диагноз болезни НП-С (например, проявляющейся холестатическим поражением печени у новорожденных).
Патогномонично для НП-С обнаружение полиморфных цитоплазматических включений при электронной микроскопии биоптатов печени или кожи, однако, их выявляют не всегда [5, 16, 22].
Генетическое исследование является золотым стандартом для окончательной верификации диагноза, а также для пренатальной диагностики [5, 16]. Молекулярно-генетическое исследование генов NPC1 и NPC2 проводится в специализированных лабораториях. В некоторых случаях для выявления мутации NPC1 необходимо выполнить комбинированное исследование gДНК и cДНК [5, 6, 16, 23]. Выявление носителей и пренатальный диагноз
В случае подтверждения диагноза болезни НП-С необходимо осуществить генетическое консультирование пациента: уточнить характер заболевания, тип наследования, дать рекомендации по планированию семьи (с проведением пренатальной диагностики). ДНК рекомендовано исследовать у обоих родителей.
Пренатальный диагноз может быть установлен на основании исследования хорионических ворсинок на 10–12-й нед беременности. Предпочтительным считают молекулярно-генетический анализ, для которого (в отличие от биохимической пренатальной диагностики) не требуется культура клеток [5].
Разнородность и вариабельность клинических проявлений НП-С в зависимости от возраста манифестации обусловливают гиподиагностику болезни. Это очевидно на примере нашей страны: на начало 2010 г. в РФ было диагностировано всего 2 пациента с диагнозом «болезнь Ниманна – Пика, тип С» [24], но и в последующем, несмотря на многочисленные публикации по данной теме в медицинской периодической литературе, проведение научно-практических семинаров, зарегистрировано всего 7 случаев данной патологии [2, 10, 24, 25]. Учитывая распространенность болезни, очевидно, что пациентов с болезнью НП-С в стране должно быть больше. А это означает, что они в отсутствие специфической терапии подвергаются риску быстрого прогрессирования болезни, инвалидизации и, самое худшее, более раннему наступлению неблагоприятного исхода. В то время как раннее назначение специфической терапии позволяет замедлить дальнейшее развитие болезни. В настоящее время лучшую доказательную базу по эффективности и безопасности имеет лекарственный препарат миглустат (Завеска, Actelion Pharmaceuticals Ltd, Швейцария). Это единственный препарат, разрешенный для применения при НП-С. Миглустат — небольшая молекула (иминосахар), которая конкурентно ингибирует фермент глюкозилцерамид синтетазу, катализирующую первый этап синтеза гликосфинголипидов [26–28], тем самым подавляет накопление нейротоксичных ганглиозидов GM2 и GM3, лактозилцерамида и глюкозилцерамида. В странах Евросоюза (ЕС), США и некоторых других миглустат применяют для лечения больных с легкой или среднетяжелой формами болезни Гоше 1-го типа, которым не показана заместительная терапия ферментами [26–28]. Миглустат зарегистрирован для лечения болезни НП-С в 2006 г. в странах ЕС, в 2008 г. — в США. В январе 2009 г. Комиссия ЕС расширила показания к применению миглустата: лечение прогрессирующих неврологических проявлений НП-С у взрослых и детей [2].
Поскольку время начала специфической и адекватной симптоматической терапии имеет решающее значение для прогноза при болезни НП-С, важно как можно раньше выявить болезнь — установить диагноз. Данная проблема побудила группу исследователей во главе с профессором F.A. Wijburg к созданию алгоритма диагностики болезни НП-С [29].
Ученые поставили перед собой следующие цели:
1) разработать прогностический инструмент «индекс подозрения болезни НП-С» и валидировать специфичность и чувствительность показателей на основании анализа историй болезней пациентов; 2) разработать балльную шкалу оценки симптомов и их комбинаций для оценки вероятности диагноза болезни НП-С; 3) разработать простой инструмент, который мог бы позволить врачам различных специальностей, не знакомым с проблемой болезни НП-С: — понимать основные симптомы и проявления, — предположить у пациента болезнь НП-С как возможный диагноз.
В рамках данного исследования все симптомы болезни НП-С были разделены на три основные категории: висцеральные, неврологические и психиатрические. Ретроспективный анализ данных 216 пациентов, направленных на обследование с подозрением на болезнь НП-С в 7 центров Европы и Австралии, позволил оценить диагностическую важность каждого отдельного симптома и их совокупности. По результатам статистического анализа, симптомы внутри каждой категории были разделены на 5 групп по степени вероятности болезни НП-С: от наиболее вероятных (40 баллов/пункт), таких как вертикальный супрануклеарный паралич взора и геластическая катаплексия, до маловероятных или дополнительных (1 балл/пункт), таких как гипотония, судороги и миоклонус. К примеру, атаксия встречается примерно у 80% пациентов с болезнью Ниманна – Пика, тип С [1], как симптом в данном алгоритме оценена в 10 баллов (средний индекс вероятности). Подобный симптом может встречаться при многих других наследственных болезнях, то есть специфичность его невысока. В то же время необъяснимая изолированная спленомегалия, встречающаяся примерно в 50% случаев болезни НП-С [20], является высокоспецифичным симптомом этой патологии (20 баллов).
В разделе «висцеральные симптомы» учитываются также данные анамнеза. Несмотря на возможность полного разрешения в течение первого года жизни затяжной желтухи, гепатоспленомегалии (в случае их наличия у пациентов в периоде новорожденности), эти симптомы являются важными для постановки диагноза.
В ходе разработки диагностического алгоритма была проведена оценка взаимосвязи симптомов. Наличие у пациента одновременно симптомов из категорий «неврологические» и «висцеральные» повышает прогностический балл НП-С на 40, как и сочетание висцеральных и психиатрических симптомов. Сочетание неврологических и психиатрических симптомов оценено в 20 баллов. Дополнительно оценивается семейный риск: 40 баллов — при наличии родных братьев/сестер или родителей с болезнью Ниманна – Пика, тип С, 20 баллов — двоюродных братьев/сестер с данной патологией.
Суммарная оценка или прогностический балл позволяет врачу разработать тактику дальнейшего ведения пациента:
<40 баллов — диагноз болезни НП-С маловероятен; 40–69 баллов — необходимо дальнейшее наблюдение, консультация в специализированном центре для проведения дифференциальной диагностики; ≥70 баллов — высокая вероятность болезни НП-С, необходимо срочно направить пациента в специализированный центр для диагностики.
Список литературы
1. Vanier M.T., Millat G. Niemann–Pick disease type C. Clin. Genet. 2003; 64: 269–281. 2. Wraith J, Baumgartner M, Bembi B. Рекомендации по диагностике и лечению болезни Ниманна – Пика типа С. Педиатрическая фармакология. 2010; 7 (1): 16–24. 3. URL: https://www.esgld.org/ (European Study Group on Lysosomal Diseases — ESGLD). 4. URL: https://www.hgmd.cf.ac.uk HGMD (Human Gene Mutation Database, Cardiff). 5. Vanier MT. Prenatal diagnosis of Niemann–Pick diseases types A, B and C. Prenat. Diagn. 2002; 22: 630–632. 6. Millat G, Bailo N, Molinero S et al. Niemann–Pick C disease: use of denaturing high performance liquid chromatography for the detection of NPC1 and NPC2 genetic variations and impact on management of patients and families. Mol. Genet. Metab. 2005; 86: 220–232. 7. Subramanian K, Balch WE. NPC1/NPC2 function as a tag team duo to mobilize cholesterol. Proc. Natl. Acad. Sci. 2008; 105: 15223–15224. 8. Sleat DE, Wiseman JA, El-Banna M et al. Genetic evidence for nonredundant functional cooperativity between NPC1 and NPC2 in lipid transport. Proc. Natl. Acad. Sci. 2004; 101: 5886–5891. 9. Fernandez-Valero E, Ballart A, Itturiaga C et al. Identification of 25 new mutations in 40 unrelated Spanish Niemann–Pick type C patients: genotype-phenotype correlations. Clin. Genet. 2005; 68: 245–254. 10. Михайлова С.В., Захарова Е.Ю., Букина Т.М. и др. Болезнь Ниманна – Пика тип С: Клинические примеры. Педиатрическая фармакология. 2010; 7 (5): 48–53. 11. Scriver CR et al. The Metabolic and molecular bases of inherited diseases (McGraw-Hill). New York, 2005. 12. Imrie J, Dasgupta S, Besley GT et al. The natural history of Niemann–Pick disease type C in the UK. J. Inherit. Metab. Dis. 2007; 30: 51–59. 13. Kelly DA, Portmann B, Mowat AP et al. Niemann–Pick disease type C: diagnosis and outcome in children, with particular reference to liver disease. J. Pediatr. 1993; 123: 242–247. 14. Solomon D, Winkelman AC, Zee DS et al. Niemann–Pick type C disease in two affected sisters: ocular motor recordings and brainstem neuropathology. Ann. N. Y. Acad. Sci. 2005; 1039: 436–445. 15. Zafeiriou DI, Triantafyllou P, Gombakis NP et al. Niemann–Pick type C disease associated with peripheral neuropathy. Pediatr. Neurol. 2003; 29: 242–244. 16. Patterson MC, Vanier MT, Suzuki K et al. The Metabolic and molecular bases of inherited disease (McGraw-Hill). New York, 2001. 3611–3633. 17. Sevin M, Lesca G, Baumann N et al. The adult form of Niemann–Pick disease type C. Brain. 2007; 130: 120–133. 18. Bauer P, Boettcher T, Meyer WP et al. Niemann–Pick type C disease (NP-C) is a considerable diagnosis in juvenile and adult-onset psychiatric disorders. Annual Meeting of theAmerican Society of Human Genetics (ASH-G), Philadelphia, USA, 2008. 19. Argoff CE, Comly ME, Blanchette-Mackie J et al. Type C Niemann–Pick disease: cellular uncoupling of cholesterol homeostasis is linked to the severity of disruption in the intracellular transport of exogenously derived cholesterol. Biochim. Biophys. Acta. 1991; 1096: 319–327. 20. Vanier MT, Rodriguez-Lafrasse C, Rousson R et al. Type C Niemann–Pick disease: spectrum of phenotypic variation in disruption of intracellular LDL-derived cholesterol processing. Biochim. Biophys. Acta. 1991; 1096: 328–337. 21. Strauss JF, Liu P, Christenson LK et al. Sterols and intracellular vesicular trafficking: lessons from the study of NPC1. Steroids. 2002; 67: 947–51. 22. Vanier MT, Millat G. Structure and function of the NPC2 protein . Biochim. Biophys. Acta. 2004; 1685: 14–21. 23. Paciorkowski AR, Westwell M, Ounpuu S et al. Motion analysis of a child with Niemann–Pick disease type C treated with miglustat. Mov. Disord. 2008; 23: 124–128. 24. Руденская Г.Е., Захарова Е.Ю., Букина Т.М. и др. Болезнь Нимана – Пика, тип С (ювенильный дистонический липидоз). Журн. неврол. и психиатр. им. С.С. Корсакова. 2008; 5: 76–79. 25. Болезнь Ниманна – Пика тип С глазами матери, медицинской сестры, врача. Педиатрическая фармакология. 2010; 7 (2): 149–150. 26. Butters TD, Dwek RA, Platt FM. Inhibition of glycosphingolipid biosynthesis: application to lysosomal storage disorders. Chem. Rev. 2000; 100: 4683–4696. 27. Wraith JE, Vecchio D, Jacklin E et al. Disease stability in patients with Niemann–Pick disease type C treated with miglustat, Lysosomal diseases network WORLD symposium 2009. San Diego, California, USA, 2009. 28. Wraith JE, Pineda M, Sedel F et al. Miglustat in patients with Niemann–Pick Type C disease (NP-C): a multicentre retrospective survey. Annual Meeting of the American Society of Human Genetics, Philadelphia, USA, 2008. 29. Wijburg FA et al. NP-C disease, an autosomal recessive neurovisceral cellular lipidosis, is characterized by the inappropriate movement and partitioning of LDL-derived cholesterol. Cell. Res. 2005; 255: 56–66.
Диагностика заболевания
Как правило, типичные симптомы заболевания можно заметить уже в первые месяцы жизни малыша. Увеличение печени и селезенки, безразличие к окружающей среде и вялость являются первыми признаками патологии, при выявлении которых ребенка направляют на анализы в центр генетики. Ныне используется целый комплекс методик, которые позволяют не только установить диагноз, но и определить тип болезни, сделать прогнозы.
- В культуре клеток кожи выявляют дефекты 11, 14 или 18 пар генов;
- проводят анализ активности сфингомиелиназы в культуре лейкоцитов;
- наиболее точные результаты получают при биопсии костного мозга, в котором выявляются характерные «пенистые» клетки.
Лечение болезни Ниманна-Пика за границей не начинают без проведения дополнительной диагностики, необходимой для определения степени анатомических изменений в органах и тканях. Обязательно проводится компьютерная томография головного мозга и УЗИ брюшной полости, психиатрические тесты.
Этиология развития патологии Нимана-Пика
Основная причина развитие патологии Нимана-Пика — это дефект в хромосомах:
- В хромосоме №11 (тип патологии А);
- В хромосоме №14 и №18 (тип патологии В).
При нарушении и дефекте в хромосомах, происходит снижение активности молекул сфингомиелиназы, которая расщепляет молекулы жиров сфингомиелина.
При таком нарушении в активности, происходит накапливания молекул жира сфингомиелина совместно с молекулами холестерина в макрофагах, что приводит к нарушению липидного обмена, а также к метаболизму всего организма.
Факторы-провокаторы, которые могут усугубить прогрессирование заболевания Нимана-Пика:
- Неправильное питание с преобладанием в рационе животных жиров;
- Злоупотребление алкоголем;
- Гиподинамия и полностью отсутствуют нагрузки на организм;
- Избыточный вес — ожирение;
- Постоянное перенапряжение нервной системы;
- Частые стрессовые ситуации;
- Хронические патологии в организме.
Если происходит мутирование нескольких генов одновременно, тогда болезнь Нимана-Пика протекает в осложнённой форме.
При нарушении и дефекте в хромосомах, происходит снижение активности молекул сфингомиелиназы
Лечение патологии в Германии
На протяжении долгих лет для лечения болезни Ниманна-Пика применялся лишь ряд медикаментозных средств, сглаживающих симптоматику болезни. При хроническом течении типов B и C подобный подход позволял существенно улучшить качество жизни пациента и продлить ему жизнь.
Сегодня лечение болезни Ниманна-Пика в Германии основано на последних достижениях медицины и генетики. Широко применяются препараты, блокирующие синтез сфингомиелина, накопление которого и является причиной увеличения внутренних органов и психических расстройств. Их применение показано пациентам на протяжении всей жизни.
Высокие результаты дает трансплантация костного мозга, при которой стволовые клетки с дефектным генотипом заменяются на здоровые.
Сроки проведения трансплантации определяются не столько ценой, сколько временем поиска подходящего донора. Упрощает задачу наличие здоровых родственников с подходящим набором иммуноглобулинов. При значительном увеличении органов проводят операцию по удалению селезенки и резекции печени.
Цены на лечение болезни Ниманна-Пика в Германии варьируются в зависимости от количества процедур и необходимых операций. Доброкачественное течение по типу B крайне редко требует хирургического решения, в то время как тип А требует неотложного квалифицированного вмешательства.
Наследственные нервно-мышечные заболевания
Наследственные нервно-мышечные болезни представлены самой обширной группой патологий.
К 1 группе нервно-мышечной дистрофии относятся спинальные амиотрофии. К распространенным формам спинальной амиотрофии относятся:
- прогрессирующая спинальная амиотрофия Арана-Дюшенна, наблюдающаяся у взрослых;
- юношеская амиотрофия Кугельберга-Веландера;
- детская форма амиотрофии Верднига-Гоффмана.
Вторая группа нервно-мышечной дистрофии представлена невральными амиотрофиями. Эти заболевания могут быть как семейными, так и спорадическими. Эта группа заболеваний, как правило, проявляется в детском или юношеском возрасте. К этой группе нервно-мышечных дистрофий относятся:
- синдром Русси-Леви;
- невральная форма амиотрофии Шарко-Мари-Тута;
- болезнь Рефсума;
- гипертрофическая интерстициальная невропатия Дежерина-Сотта.
Обширная группа нервно-мышечных заболеваний представлена первичными прогрессирующими мышечными дистрофиями. Проявления первичных прогрессирующих мышечных дистрофий могут быть разнообразными. Яркими представителями этой группы заболеваний являются:
- псевдогипертрофическая детская дистрофия Дюшенна;
- благоприятно текущая псевдогипертрофическая дистрофия Беккера-Кинера;
- ювенальная и конечно-поясничная дистрофия Эрба;
- дистальная мышечная дистрофия;
- окулофарингеальная и окулярная форма дистрофии;
- синдром ригидного позвоночника;
- непрогрессирующая мышечная дистрофия.
- наследственная парамиотония Эйленбурга;
- миотония Томсена;
- нейромиотония;
- дистрофическая миотония.
К 5 группе нервно-мышечных дистрофий относятся миоплегические синдромы и пароксизмальные миоплегии. К самым распространенным миоплегиям относятся:
- нормокалиемическая миоплегия;
- болезнь Гамсторп;
- гипокалиемическая пароксизмальная миоплегия;
- вторичные варианты пароксизмальной миоплегии.
Все эти заболеваниям имеют свои особенности течения и развития. Некоторые из них позволяют больным жить полноценной жизнью и не испытывать явного дискомфорта, в то время как другие приводят к значительным нарушениям работы мышечного аппарата.
DMU: комплексное сопровождение лечения в Германии
Тяжелые синдромы и угроза жизни ребенка при болезни Ниманна-Пика диктуют родителям свои условия – лечение должно быть оказано вне зависимости от его цены, качественно и без отлагательства. Если при этом Вас останавливает сложный выбор и поиск клиники, отсутствие знаний языка, страх перед перелетами, необходимость размещения в чужой стране и сложность при оформлении виз, наши специалисты готовы помочь и взять под свою ответственность все организационные вопросы. Ведь болезнь Ниманна-Пика в Германии рассматривается и лечится комплексно, что позволяет компенсировать патологию в любом возрасте.
Специалисты центра DMU в России будут рядом с Вами на протяжении всего цикла лечения:
- мы подберем для Вас лучших специалистов в Германии, предоставим смету, план лечения и назначим визит;
- поможем с оформлением медицинской визы;
- забронируем билеты и будем сопровождать Вас от отъезда до возвращения домой;
- рядом с вами будет профессиональный переводчик, который разъяснит все, что говорит врач.
Вы можете быть уверены и в обосновании цен на каждую услугу. Наши координаторы тщательно проверяют выставленные счета и сверяют их с рекомендованным Минздравом Германии прейскурантом. Лечение за рубежом может стать Вашим билетом в новую жизнь, и мы готовы поддержать Вас в каждом действии.
Классификация
Различают три клинические формы данного недуга:
- тип А — классическая инфантильная форма. Симптоматика проявляется уже на первом году жизни, характерны нарушения неврологического характера (судороги, затруднённое глотание, отсутствие реакции). В большинстве случаев дети умирают до 3-х лет;
- тип В – висцеральная форма. Симптоматика может проявляться в возрасте от 2 до 6 лет. Больше всего поражается печень и селезёнка. Риск летального исхода намного ниже, многие больные доживают до преклонного возраста;
- тип С — подростковая неострая форма. Симптоматика проявляется в возрасте 2–5 лет, более интенсивной становится в 15–18 лет. Поражаются внутренние органы, в том числе и головной мозг. Чаще всего пациенты умирают в 15–18 лет.
Наиболее благоприятной считается висцеральная форма заболевания, так как летальный исход диагностируется крайне редко, а клиническая картина менее выражена.
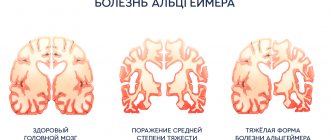
Каковы прогнозы?
К сожалению, прогнозы для пациентов с подобным диагнозом неутешительные. Болезнь Пика — проблема крайне серьезная, и на сегодняшний день не существует способов остановить разрушение нейронов. Тем не менее использование подходящих препаратов, качественная реабилитация помогают замедлить процесс, сохраняя качество жизни пациента.
Но, согласно статистике, в течение 5 – 10 лет болезнь приводит к психическому и моральному разложению личности — у пациентов развивается слабоумие, маразм, кахексия и т.д. Такие больные должны находиться под постоянным присмотром, желательно — в специализированных клиниках, где за ними будут постоянно следить медицинские сотрудники.
Существует ли эффективное лечение?
Какой терапии требует болезнь Пика? Лечение на сегодняшний день сводится к попыткам приостановить дальнейшее прогрессирование заболевания. В первую очередь больным необходима заместительная терапия — пациентам назначают прием тех веществ, синтез которых в организме не происходит из-за атрофии нервных тканей (например, ингибиторы ацетилхолинестеразы). В некоторых случаях необходим дополнительный прием противовоспалительных веществ.
Крайне важной частью терапии является и прием нейропротекторов, которые стимулируют процесс жизнедеятельности нейронов. При наличии психических расстройств используются соответственные препараты, которые помогают ослабить симптомы, например, устранить агрессию. Иногда необходим прием антидепрессантов.
Безусловно, важной частью лечения является психологическая поддержка. Пациентам рекомендуют регулярно посещать специальные занятия и тренинги, которые помогают приостановить процесс психических и эмоциональных изменений.
Какими симптомами сопровождается дальнейшее развитие болезни?
По мере развития болезни симптомы становятся все более выраженными. Нередко у человека наблюдаются расстройства речи — словарный запас становится более скудным и со временем сводится к нескольким фразам, которые пациент повторяет постоянно. Естественно, подобное сказывается и на письме. К признакам болезни Пика можно отнести афазию, алексию, аграфию и т.д.
Кроме того, в некоторых случаях заболевание сопровождается появлением гипокинезов, потерей мышечного тонуса и т.д. Довольно часто пациенты с подобным диагнозом страдают от ожирения, а также отказываются соблюдать правила личной гигиены.
В любом случае на более поздних этапах пациенту необходима помощь — человек больше не в состоянии сам о себе заботиться.