1.General information
Neurodegeneration is a pathological process, with the development of which nervous tissue loses its complex organization, degenerates, gradually atrophies (decreases in volume and dies), becoming, in general, functionally untenable. Considering that the nervous system controls and regulates literally everything in the body, neurodegenerative diseases, even the slowest and most indolent ones, always constitute a serious problem, which is aggravated by the fact that at the moment all efforts to develop reparative (restorative) and etiopathogenetic ( eliminating the root cause of the disease) types of therapy have not brought tangible results.
For the most part, neurodegenerative diseases are caused by, or at least reliably associated with, hereditary, chromosomal factors. These diseases are traditionally considered rare, and in terms of tens and hundreds of thousands of people, many of them really seem to be sporadic, almost random anomalies. However, if we sum up the incidence of the fairly well-known diseases Alzheimer's, Pick's, Parkinson's, demyelinating multiple or amyotrophic lateral sclerosis, and DLB (dementia with Lewy bodies), the picture will look more alarming. Thus, with reference to data from post-mortem pathomorphological studies, it has been repeatedly emphasized in the literature that the same DLB (one of the frontotemporal variants of neurodegeneration) is diagnosed much less frequently than it actually occurs.
A large number of individual nosological units (i.e., officially established diagnoses of this group) are the subject of critical discussions, since degeneration of neural tissue is the main and common mechanism for the development of such diseases; for example, the extreme wing of supporters of generalization proposed “for convenience” to consider all diseases of this kind as only particular variants of Alzheimer’s or Parkinson’s diseases. This approach can hardly be considered justified: the clinical picture, rate of progression, prognosis, strategy of symptomatic treatment - all this is determined by a number of significant factors (primarily the predominant localization of the process) and differs sufficiently to speak of independent diseases.
The above fully applies to spinocerebellar ataxia. This is a hereditary neurodegenerative disease with its own distinct specificity, which can manifest at any age (usually in the range of 5-40 years) and is distinguished by a variety of forms: to date, over twenty relatively independent types have been identified and described (SCA, SCA 2, Friedreich's disease and many others). etc.).
A must read! Help with treatment and hospitalization!
Treatment in Italy
Russian Medical Server / Treatment in Italy / Center for the treatment of rare diseases in Milan / Spinocerebellar ataxia - treatment in Italy
Spinocerebellar ataxia includes various actively developing hereditary forms of pathologies, characterized by impaired coordination of movements. With such lesions of the human body, the main changes occur in the cerebellum, in the spinal cord, and also in the brain stem. Among all known hereditary diseases, spinocerebellar ataxias are in second place in frequency of occurrence after neuromuscular pathologies.
The disease is characterized by a noticeable clinical diversity of symptoms; there is a wide gradation between mixed forms of pathology and cerebellar types. There are many different classifications based on clinical and anatomical principles and on the types of genetic inheritance of disease symptoms.
It is important to know that all known classifications of spinocerebellar degenerative processes are not able to fully satisfy the requirements of medicine. This can mainly be explained by insufficient study of the disease and the lack of basic understanding of the development of biochemical defects that become causative.
Spinocerebral ataxia occurs with a frequency in different populations of approximately 1 - 23 cases per hundred thousand population. The pathology has an uneven ethnic and geographic distribution. In Russia and Italy, the first type of disease mainly predominates; in India, the second type of spinocerebellar ataxia is more common; the third type of disease is most often diagnosed in the population of Germany, the USA and Japan.
The group of spinocerebellar ataxias refers to cerebellar lesions that are inherited in an autosomal dominant manner. At the same time, mutations occurring in genes provoke the formation of pathological protein products that cause the death and death of cells in the cerebellum, spinal cord, and cerebral cortex.
The neurological symptoms of spinocerebellar ataxia develop rather slowly, and the development itself can be delayed for up to twenty years, but the disease can also progress more quickly. Sometimes there are periods of stable conditions.
With the development of concomitant infectious lesions, additional symptoms begin to appear. Patients with very advanced stages of pathology are deprived of the ability to get out of bed, they develop dysphagia, that is, a violation of the act of swallowing, and other similar signs. A person may die due to exhaustion and often from the development of myocarditis, accompanied by severe forms of heart failure. With proper care, the patient lives up to forty to fifty years.
Subsequently, the symptoms are supplemented by tremors of the legs and arms when performing any actions and when coordination of movements is impaired. In addition, handwriting begins to change - it becomes uneven, and the letters are too large, and speech also changes.
The disease is also characterized by eye movement disorders - jerky, sharp movements of the eyeballs when the gaze shifts from one object to another. Often there are disturbances in swallowing, speech pronunciation, disruption of the hearing aid, disturbances in stool and urine output, paralysis of the legs and arms, and pathological reflexes develop. And habitual reflexes simultaneously worsen reactions and then disappear completely.
At times, the disease occurs in an abortive or mild form and provokes minor disability of the patient or is not accompanied by disability at all. Such pathologies can be diagnosed in relatives of the patient who suffer from advanced clinical types of the disease.
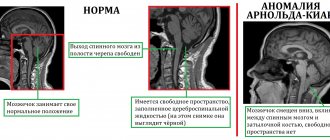
When diagnosing, preference is given to imaging methods such as computed tomography or magnetic resonance imaging. They make it possible to identify areas of degeneration of nerve fibers, demyelination of pons neurons, expansion of all components of the cerebrospinal fluid circulation system in the brain, and atrophic changes in the cerebral cortex.
To make a diagnosis, it is also necessary to exclude some other diseases that occur with a similar clinical picture (such as tumors of the posterior cranial fossa, multiple sclerosis, hydrocephalus, vascular pathology of the brain, especially in the spinobasilar region. If, based on the clinical picture, it is possible to confirm the development of ataxia, then it is much more difficult to determine the type of disease (if there are no strictly specific symptoms).
Currently, there are several opinions regarding the treatment tactics for this pathology. Supporters of the first are of the opinion that ataxia is not susceptible to drug treatment. In this case, it is best to carry out maintenance therapy to slow the progression of the disease. The mandatory complex includes exercise therapy (vestibular training), as well as many methods of social, household and work rehabilitation.
Another theory involves the use of certain drugs, however, the treatment is purely symptomatic or stimulating. Injections of vitamins are prescribed, as well as their oral administration. As an element of such therapy, it is possible to prescribe anticonvulsants (if indicated). Treatment of spinocerebellar ataxia is also supported by certain physiotherapeutic procedures (for example, electrical stimulation).
! Despite the fact that many of the diseases described in this section are considered incurable, the Center for the Treatment of Rare Diseases in Milan is constantly looking for new methods. Thanks to gene therapy, it has been possible to achieve outstanding results and completely cure some rare syndromes.
Contact a consultant on the website or leave a request - this way you can find out what methods Italian doctors offer. Perhaps this disease has already been treated in Milan.
+7 (925) 50 254 50 – urgent treatment in Italy
REQUEST TO THE CLINIC
2. Reasons
Spinocerebellar ataxia is based on an inherited mutation of certain genes (the type of inheritance, as a rule, is such that if both parents are carriers, then the probability of the pathology “triggering” in the child is 1/4 or 25%). As a result, a number of complex electrochemical processes that control energy and protein balance are disrupted (the defective structure of the frataxin protein plays a significant role), the transmission of nerve impulses from the center to the periphery and back, etc. The unifying feature for the entire group of ataxias is that in neural degeneration structures of both the brain (primarily the cerebellum) and the spinal cord are involved, as well as the pathways between them, peripheral nerves and, in some cases, myocardial tissue. The term “ataxia” literally means “lack of order, coherence,” and, by definition, the main manifestation of spinocerebellar ataxia is progressive impairment of motor coordination and, in general, neuromuscular coherence.
Visit our Neurology page
Spinocerebellar ataxias
Despite the significant genetic and partly clinical diversity of spinocerebellar ataxias, the molecular mechanisms of genetic disorders in these diseases are very similar. The main cause of the pathology is a change in the number of trinucleotide sequences (CAG) in the coding part of the disease-associated genes. This leads to an increase in the amount of the amino acid glutamine in the resulting protein, which changes the physicochemical properties of the protein and impairs its functions. In some cases, the above proteins directly or indirectly participate in the metabolism of nervous tissue, so changes in their structure lead to spinocerebellar ataxia. Currently, the molecular mechanisms of the 6 main types of this disease are best studied - these forms of pathology are the most common and together account for more than 90% of cases of spinocerebellar ataxia.
Spinocerebellar ataxia type 1
is considered the most common and most studied variant of this pathology. It is caused by mutations in the ATXN1 gene, which is located on chromosome 6. Normally, this gene has no more than 36 CAG repeats; an increase in their number leads to the development of the disease. The product of expression of the ATXN1 gene is a special DNA-binding protein that is actively involved in the metabolism of Purkinje cells of the cerebellum - in the presence of a mutant version of the gene, this leads to the appearance of aggregates and gradual degeneration, which becomes the cause of spinocerebellar ataxia.
Spinocerebellar ataxia type 2
– a less common variant of the disease, the etiology has not been studied so thoroughly. The cause of the pathology is an increase in the number of CAG repeats in the ATXN2 gene, localized on chromosome 12. In a healthy version of the gene, the number of the above sequences ranges from 15 to 36, while in spinocerebellar ataxia there can be over 100. The functions of the protein encoded by the ATXN2 gene are currently unknown.
Spinocerebellar ataxia type 3
(another name is Machado-Joseph disease in honor of the two patients in whom this condition was first described) - the cause of this variant of pathology is disturbances in the ATXN3 gene, located on the 14th chromosome. Normally, the number of CAG repeats in this gene does not exceed 47; with the development of the disease, from 53 to 68 repeats are detected. This gene encodes a protein that is presumably involved in the energy metabolism of neurons in the cerebellum and basal ganglia.
Spinocerebellar ataxia type 6
– a relatively rare type of disease caused by defects in the CACNA1A gene, localized on the 19th chromosome. For the development of pathology, a very slight increase in the number of CAG repeats is sufficient - if in a normal variant of the gene they are found in 5-20, then in the presence of ataxia - 21-26. The CACNA1A gene encodes a protein subunit of calcium channels located on cerebellar neurons. In addition to spinocerebellar ataxia, disturbances in the CACNA1A gene cause the development of episodic ataxia and some hereditary forms of migraine.
Spinocerebellar ataxia type 7
– this type of pathology is caused by disturbances in the structure of the ATXN7 gene, which is located on the 3rd chromosome. In a healthy person, the number of CAG repeats is no more than 35, while in a disease their number can reach several hundred. The functions of the protein that encodes the ATXN7 gene are currently being studied.
Spinocerebellar ataxia type 8
is caused by a genetic defect in the ATXN8 gene, located on chromosome 13. As in other cases, the essence of the genetic defect in this condition is a change in the number of CAG trinucleotide sequences - usually there are about 15-50, while in pathology the number of repeats can be over 1200.
In almost any type of spinocerebellar ataxia, an abnormal form of the protein, excessively rich in glutamine, forms deposits in the nuclei or cytoplasm of neurons of the cerebellum and basal ganglia in the form of dense aggregates. This process goes the faster, the more the number of CAG repeats in the key gene differs from the norm. This also explains the mechanism of anticipation of the symptoms of spinocerebellar ataxia - during the process of meiosis during the formation of germ cells, the number of the above trinucleotide sequences can increase, which leads to increased symptoms.
Since a similar phenomenon more often occurs during the formation of male germ cells, this becomes the cause of the so-called “paternal transmission”, when anticipation is recorded only when the disease is transmitted from the father to the offspring. Many geneticists believe that the main cause of spinocerebellar ataxia lies not in the increase in “histidine” trinucleotides, but in the deletion of the so-called regulatory triplets that separate the CAG repeat regions. For example, in the first type of disease it is CAT, in the second CAA - they regulate the number of CAG repeats and maintain the stability of their number during meiosis.
3. Symptoms, diagnosis
The probable symptoms of spinocerebellar neuronal degeneration are so polymorphic that it is impossible to describe at least the main of its twenty types in one article. There are disorders of visual-motor coordination and muscle tone (tremor, extrapyramidal “stiffness”); partial paralysis of the extraocular muscles and degenerative retinopathy in combination with optic nerve atrophy; general atrophy of muscle fibers; various neurological symptom complexes. If the neurodegenerative process affects the cerebral cortex, dementia gradually develops - a debilitating decline in cognitive functions (memory, attention, various types of recognition, etc.), logical thinking, and speech organization. In case of damage to the medulla oblongata, “bulbar” symptoms develop: degradation of swallowing, palatal, chewing, and respiratory reflexes.
Diseases in this group progress relatively slowly: the course can take up to 20 years or more, although much faster developments have also been described. Patients gradually lose the ability for self-care and, in general, for productive contact with the world; they increasingly depend on the care and support of others, falling into complete helplessness towards the terminal stage and dying, as a rule, from pneumonia, exhaustion, respiratory failure, etc.
Neurodegenerative diseases, including ataxia, are diagnosed clinically, during a neurological examination and a thorough analysis of complaints and anamnestic information. Additionally, tomographic imaging methods, neuropsychological examination, etc. may be prescribed, but the final diagnosis is established and differentiated, as a rule, only pathomorphologically.
About our clinic Chistye Prudy metro station Medintercom page!
Spinocerebellar ataxia type 1 (SCA 1) is a severe neurodegenerative progressive disease with a late age of manifestation, inherited in an autosomal dominant manner; clinically characterized by a combination of increasing motor coordination disorders with signs of multisystem damage to the brain and spinal cord (Illarioshkin et al., 2002). The onset of symptoms in SCA1 is usually observed in adulthood, on average at 30 years of age. If symptoms appear earlier—before age 20—other symptoms often occur in addition to ataxia. In very early cases (before 13 years of age), the disease may be more severe and progress rapidly.
| Main clinical symptoms: The first symptom is usually poor hand coordination and poor balance when walking. In fact, the word ataxia itself means a lack of coordination. As SCA1 progresses over several years, difficulty swallowing and unclear speech occur. In some cases, patients develop additional symptoms such as neuropathy (loss of feeling and reflexes in the legs), muscle spasticity, weakness, or memory loss. |
In SCA1, genetic defects lead to deterioration in the functioning of certain nerve fibers that carry information to and from the brain, resulting in degeneration of the cerebellum (the focal point of the brain).
Parental consanguinity is often noted. The disease cannot be treated, and death most often occurs 10-15 years after the first symptoms appear. The genetic cause of SCA1 development is the expansion of the copy number of tandem trinucleotide CAG repeats in the coding region of the SCA1 gene (Orr et al., 1993). For SCA1, direct DNA diagnosis of the disease is possible at the presymptomatic stage, sometimes many years before the onset of any neurological and/or mental disorders. This is of particular importance for medical genetic counseling, since currently no effective methods for treating SCA1 have been found, although research in this direction is actively being conducted around the world (Ogawa, 2004; Dueñas et al., 2006; Takei et al., 2007; Gao et al., 2008). The only way to combat this disease today is to prevent the emergence of new cases of SCA1 in affected families (Illarioshkin et al., 1997). Until the mid-70s in Yakutia, the disease was classified as the “cerebellar form” of Vilyui encephalomyelitis. Currently, spinocerebellar ataxia type I is considered as an ethnospecific hereditary disease (Puzyrev, Maksimova, 2008). The high prevalence of the disease (38.6 per 100 thousand Yakuts compared to 1-2:100 thousand in the world population) in Yakutia was assessed as a “Siberian hotbed” of disease accumulation, the largest in the world. Areas of high accumulation of NCA1 - Abyisky and Ust-Aldan uluses of Yakutia - are characterized by a homogeneous national composition; high birth rate; low level of migration. The molecular basis of the disease is an increase in the number of trinucleotide CAG repeats in Yakuts to 39-71 compared to 19-36 normally in the SCA1 gene, localized in the 6p22 - p23 region. Among the various forms of hereditary ataxia in Yakutia, SCA 1 occurs in 88.1% of all families.
Literature:
- Kucher A.N., Danilova A.JL, Koneva L.A., Maksimova N.R., Nogovitsina A.N. Genetic and demographic study of the population of the Republic of Sakha (Yakutia) // Yakut Medical Journal. — 2005. No. 2(10). — P. 4-12.
- Koneva L.A., Kucher A.N., Maksimova N.R., Puzyrev V.P. Approaches to modeling the prevalence of spinocerebellar ataxia type I in an isolated population // Human Genetics and Pathology: Coll. scientific works / Ed. V.P. Puzyreva. — Vol. 7. - Tomsk: Printing Manufactory, 2004. -P. 92-101.
- Koneva L.A., Kucher A.N., Puzyrev V.P., Nogovitsyna A.N., Maksimova N.R., Sukhomyasova A.L., Danilova A.L. Demographic and clinical-genetic features of the prevalence of spinocerebellar ataxia type I in the Ust-Aldan and Abyi uluses of the Republic of Sakha (Yakutia) / Mater, scientific and practical scientist. conf. "Current issues of preventive medicine." -Ulan-Ude. - 2005. - P. 97-100.
- Koneva L.A., Kucher A.N., Puzyrev V.P., Maksimova N.R., Nogovitsina A.N., Sukhomyasova A.L., Danilova A.L., Platonov F.A., Korotov M. .N. Characteristics of the incidence of spinocerebellar ataxia type I in the Ust-Aldan and Abyi uluses of the Republic of Sakha (Yakutia) / Mater. International scientific-practical conf. “Genetic aspects of human pathology. Problems of preserving the gene pool of the indigenous peoples of the North”, Yakutsk: NIPK “Sakhapoligraphizdat”. - 2005. - P. 99-100.
- Koneva L.A., Kucher A.N., Puzyrev V.P., Nogovitsina A.N., Maksimova N.R., Sukhomyasova A.L., Danilova A.L. Prevalence of spinocerebellar ataxia type I in the Ust-Aldan and Abyi uluses of the Republic of Sakha (Yakutia) / Mater. Final scientific-practical conf. State Research Institute of Medical Problems of the North of the Siberian Branch of the Russian Academy of Medical Sciences "Issues of preserving and developing the health of the population of the North and Siberia", Krasnoyarsk. - 2005. - P. 127-129.
- Koneva L.A., Maksimova N.R. Prevalence of spinocerebellar ataxia type I in Yakutia: problems and solutions / Mater, competition of works of young scientists “Theoretical and applied problems of medical genetics” SB RAMS, Novosibirsk. - 2004. - P.103-109.
- Koneva L.A., Maksimova N.R., Kucher A.N., Puzyrev V.P. Dynamics of the frequency of spinocerebellar ataxia type I in the Yakuts, taking into account the specifics of the population structure / Genetics in the 21st century: current state and development prospects: Mater. 3rd Congress of VOGiS. -M. - 2004. - P.28.
4.Treatment
There is currently no treatment that reverses or even stops neurodegeneration. All types of therapy practiced today are purely palliative in nature and are aimed at mitigating the most maladaptive symptoms that reduce the quality of life and dominate in a specific clinical picture. As a rule, medications are prescribed to improve neurotrophism (nutrition of nerve tissue), vitamin complexes, massage, and physical therapy to correct and/or compensate for movement disorders.
Description
Synonyms (rus): Machado-Joseph disease
Synonyms (eng): Spinocerebellar ataxia type 3, SCA3
Biomaterial: Venous blood
Indicator(s): Expansion in the ATXN3 gene
Method(s): Polymerase chain reaction (PCR)
Container type and preanalytical features: Hematology tube with EDTA, 2 ml (purple cap)
Spinocerebellar ataxia type 3 (SCA 3) is a neurodegenerative progressive genetic disease. The pathogenesis is based on the expansion of CAG trinucleotide repeats of the unstable region of the ATXN3 gene. If the number of CAG triplet repeats is less than 60, the disease is excluded. The disease is inherited in an autosomal dominant manner, the phenomenon of anticipation is characteristic, the number of repetitions is inversely correlated with the time of manifestation of the disease and directly proportional to the severity of its course. The disease is very heterogeneous and can manifest itself in 5 variants: type 1 – rigidity, spasticity, bradykinesia without ataxia; Type 2 – ataxia, damage to upper motor neurons; Type 3 – ataxia, polyneuropathy; Type 4 – L-DOPA dependent parkinsonism; Type 5 – spastic paraplegia. The study is recommended for patients with damage to upper motor neurons, ataxia, and polyneuropathy.