A sharp bilateral decrease in visual acuity may indicate the development of a dangerous pathology - Leber optic neuropathy. This disease most often affects young males. Let's consider the causes of pathology, symptoms and features of therapy in the article.
For the first time, cases of a disease in which visual acuity rapidly decreases due to atrophy of the optic nerve were described in 1871 by Theodor Leber. The scientist observed the development of pathology in young people from several related families, which indicated a hereditary cause of the disease. The mechanism of inheritance of the disease was carefully studied, so it was possible to prove that the pathology is transmitted through the maternal line mainly to men.
Until now, the nature of the disease has not been studied well enough to find effective treatments for such a disease. Currently, treatment of patients consists of maintenance therapy, but from time to time groups of scientists present new gene therapy techniques for clinical trials, which, of course, gives hope to people with a similar diagnosis.
Leber's optic neuropathy: symptoms, treatment of the disease
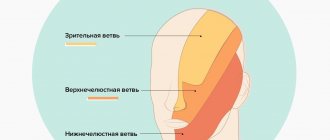
Any visual impairment that rapidly progresses must be carefully studied and comprehensively diagnosed. Leber's optic neuropathy is rarely accompanied by malaise characteristic of neurological diseases, and vision declines quickly, sometimes not even within a few months, but within two to three weeks. With such acuity, vision cannot be checked using Sivtsev’s table, because a person simply does not see the signs, so they use hardware diagnostics and the “counting fingers” method at a certain distance from the patient’s face. The peculiarity of the disease is that the optic nerve is affected due to a gene mutation. An excess amount of toxic oxygen molecules is produced, which negatively affects the condition of nerve cells and vision decreases.
Leber's optic neuropathy - symptoms and features of the disease:
- young age of patients - 18-35 years;
- the disease develops more often in men;
- rapid unilateral and then bilateral vision loss;
- visual acuity decreases over several weeks;
- violation of color perception of red and green colors;
- the pathology may be accompanied by symptoms inherent in mitochondrial diseases (convulsions, cardiac conduction disturbances).
With Leber optic nerve atrophy, most often the process of disease development begins in one eye, but there are often cases when two eyes are affected at once. Central vision is constantly decreasing. This is due to the gradual death of cells in the optic nerve, which transmits image information to the brain. Sometimes the disease may be accompanied by symptoms characteristic of neurological diseases. In this case, the sick person suffers from dystonia, tremor, and ataxia. Less often, at the beginning of the development of pathology, a person notices sparks, spots, bright flashes before the eyes.
Symptoms
The child has a lack of central vision, nystagmus, and unrecorded electroretinography. Other neurological symptoms include decreased pupillary response to light. It is quite difficult to identify this disease in a newborn for the simple reason that symptoms such as strabismus, neuromuscular and neurological disorders, hypermetropia, keratoconus, mental retardation, and poor hearing appear in many systemic diseases. The main manifestation is blindness from the birth of the child or its appearance within several months of life. Many children see better if they are given brighter lighting.
In addition, children who do not become blind early in their lives with this condition still become blind at around 10 years of age. In the first three months of life, babies are unable to respond to light or fixate on objects, and most parents notice this. They also regularly experience typical symptoms regarding eye problems in blind children, a wandering and unfocused gaze. An infant's fundus may appear completely normal, but over time, defects will begin to occur.
All patients with Leber's amaurosis aged 8-10 years have pigment deposits in the form of bone bodies on the periphery of the fundus, in some cases resembling a rubella rash. With age, rapid changes occur in the fundus of the eye, but ocular functions such as visual acuity, visual field, and ERG remain unchanged in most cases. Upon reaching the age of 15 years, a patient with Leber's congenital anauurosis is at risk of developing keratoconus.
Parents should identify this disease in their child at an early stage, since, due to his age, he is not able to complain of poor vision himself. It is necessary to monitor how he reacts and fixes his gaze on toys, whether there is anything strange in his gaze or eye movements.
Causes of Leber's hereditary optic neuropathy
The causes of Leber neuropathy, in which the optic nerve atrophies, are genetic. The disease, caused by a specific mutation in mitochondrial genes, is transmitted by women to their offspring. Men and women inherit pathology from their mother. Why is this happening? A huge amount of DNA is found in the cell nucleus and only a small part of it is in the mitochondria. Nuclear genes are inherited from both mother and father, but mitochondrial genes are inherited only from the mother. A man who inherits a mutation will not pass it on to his descendants.
In a significant proportion of people who are carriers of the mutated gene, the disease will not become apparent. More than 85% of women and 50% of men who are carriers of the LHON syndrome gene will not be affected by the disease. The reasons influencing the progression of the disease are not clear, but it is reliably known that the disease can be triggered by unfavorable environmental conditions, stress, infections, and the toxic effects of tobacco and alcohol.
Diagnosis of Leber optic neuropathy
Diagnosis of the disease is complicated by the similarity of its symptoms with ischemic neuropathy or with certain pathologies - for example, multiple sclerosis. It is also difficult to track the history, since not all carriers of the mutated gene develop Leber neuropathy. An extensive ophthalmological examination can only establish the presence of pathology, but to determine the cause, genetic diagnosis will be needed.
Features of diagnosing Leber hereditary optic neuropathy:
- general clinical examination;
- Mitochondrial DNA analysis;
- visual field examination;
- fundus examination;
- coherence tomography and electroretinography (necessary to exclude retinal pathologies).
Specialists are recommended to carry out a differential diagnosis of LHON syndrome by comparing symptoms and examination data with other diseases affecting the optic nerve. For example, with ischemic, toxic neuropathy. A complete ophthalmological examination shows that the optic disc is inflamed, and vascular telangiectasia (dilation of small capillaries) is also noticeable.
Difficulty of diagnosis
Leber's disease is difficult to diagnose. To identify this disease, the doctor must carry out a large list of diagnostic measures, which includes ophthalmoscopy, study of medical history (in particular hereditary), neuro-ophthalmological examination, and consultation with a geneticist. Often the main difficulty is to differentiate LHON from other diseases with a similar clinical picture. In particular, similar symptoms may be accompanied by neuritis in multiple sclerosis, Libman-Sachs disease and other diseases. Differential diagnosis helps to make the correct diagnosis.
Leber's hereditary optic neuropathy - can the disease be cured?
While hereditary optic neuropathy is considered an incurable disease, scientists are constantly searching for new ways to treat such a pathology. Thanks to gene therapy methods, it is possible to significantly slow down the progression of diseases and improve the quality of life of patients.
The rapid progression of Leber's disease leads to rapid loss of vision in people who have seen normally all their lives and had no vision problems. A disappointing diagnosis, as well as information that the disease has not yet been treated, seriously affects the well-being of such patients who were not prepared for disability. Many of the existing methods of treating pathology have proven to be ineffective, including surgical intervention.
Fortunately, scientists involved in rare pathologies are working hard to find a cure. Experts place particular hope in gene therapy, and many research groups have already achieved good results. Sometimes an obstacle to continuing experiments is their ethical side, since further research involves the use of the developed technique on humans. Are there any successes at least using experimental models? Yes, geneticists have already managed to find a way to solve the problem.
Thus, a group of scientists from Miami, using experimental models, proved that mutated genes can be safely replaced with healthy ones, this will prevent deterioration in the nutrition of optic nerve cells. To correct a genetic defect in the mitochondria, it is necessary to introduce normal DNA - this will correct the disorder and restore visual function. Scientists report that this approach will also be effective against other diseases caused by mitochondrial mutations, as well as various disorders associated with the aging process.
Leber's amaurosis
The main mechanism of visual impairment in Leber amaurosis is a metabolic disorder in rods and cones, which leads to lethal damage to photoreceptors and their destruction. However, the immediate cause of such changes varies depending on which gene mutation caused the disease.
One of the most common types of Leber amaurosis (type 2, LCA2) is caused by the presence of a mutant RPE65 gene on the first chromosome. More than 80 mutations of this gene are known, some of which, in addition to Leber amaurosis, also cause certain forms of retinal pigmentary abiotrophy. The protein encoded by PRE65 is responsible for the metabolism of retinol in the pigment epithelium of the retina, therefore, in the presence of a genetic defect, this process is disrupted with the development of side metabolic pathways. As a result, the synthesis of rhodopsin in photoreceptors stops, which leads to the characteristic clinical picture of the disease. Mutant forms of the gene are inherited according to an autosomal recessive mechanism.
A less common form of Leber amaurosis (type 14) is caused by a mutation in the LRAT gene on chromosome 4. It encodes the lecithin retinol acyltransferase protein, which is located in the microsomes of hepatocytes and is found in the retina. This enzyme is involved in the metabolism of retinoids and vitamin A; due to the presence of mutations in the gene, the resulting protein cannot fully perform its functions, which is why photoreceptor degeneration develops, which is clinically manifested by Leber amaurosis or juvenile retinal pigment abiotrophy. It has an autosomal recessive inheritance pattern.
Leber amaurosis type 8 most often leads to congenital blindness; the CRB1 gene responsible for the development of this form of the disease is located on chromosome 1 and has an autosomal recessive inheritance pattern. It was found that the protein encoded by this gene is directly involved in the embryonic development of photoreceptors and retinal pigment epithelium. More accurate data on the pathogenesis of this form of Leber amaurosis have not been accumulated to date. The situation is similar with the mutation of the LCA5 gene, located on chromosome 6 and associated with type 5 amaurosis. Currently, the only protein encoded by this gene, lebercillin, has been identified, but its functions in the retina are unclear.
Two forms of Leber amaurosis have also been identified, which are inherited by an autosomal dominant mechanism - type 7, caused by a mutation of the CRX gene, and type 11, associated with a disorder of the IMPDH1 gene. The CRX gene encodes a protein that has many functions - controlling the development of photoreceptors in the embryonic period, maintaining their adequate level in adulthood, participating in the synthesis of other retinal proteins (it is a transcription factor). Therefore, depending on the nature of the CRX gene mutation, the clinical picture of Leber amaurosis type 7 can be varied - from congenital blindness to relatively late and sluggish visual impairment.
Inosine 5′-monophosphate dehydrogenase 1, encoded by the IMPDH1 gene, is an enzyme that regulates cell growth and nucleic acid production, but it does not yet provide insight into the pathogenesis of how abnormalities in this protein lead to Leber amaurosis type 11.