1.Общие сведения
Нейродегенерация – патологический процесс, с развитием которого нервная ткань утрачивает свою сложнейшую организацию, вырождается, постепенно атрофируется (уменьшается в объеме и отмирает), становясь, в целом, функционально несостоятельной. Учитывая, что нервная система контролирует и регулирует в организме буквально всё, нейродегенеративные заболевания, – даже самые медленные и вялотекущие, – всегда составляют серьезную проблему, которая усугубляется еще и тем, что на данный момент все усилия по разработке репаративных (восстановительных) и этиопатогенетических (устраняющих первопричину болезни) видов терапии не принесли ощутимых результатов.
В большинстве своем нейродегенеративные болезни обусловлены или, по крайней мере, достоверно связаны с наследственными, хромосомными факторами. Эти заболевания традиционно считаются редкими, и в пересчете на десятки и сотни тысяч населения многие из них действительно кажутся спорадическими, почти случайными аномалиями. Однако если просуммировать частоту встречаемости достаточно известных болезней Альцгеймера, Пика, Паркинсона, демиелинизирующего рассеянного или бокового амиотрофического склероза, ДТЛ (деменция с тельцами Леви), картина будет выглядеть более тревожной. Так, со ссылкой на данные посмертных патоморфологических исследований в литературе неоднократно подчеркивалось, что та же ДТЛ (один из лобно-височных вариантов нейродегенерации) диагностируется значительно реже, чем в действительности встречается.
Большое число отдельных нозологических единиц (т.е. официально устанавливаемых диагнозов этой группы) служит предметом критических дискуссий, поскольку дегенерация нейронной ткани является основным и общим механизмом развития таких болезней; напр., крайнее крыло сторонников обобщения предлагало «для удобства» считать все заболевания такого рода лишь частными вариантами болезней Альцгеймера или Паркинсона. Едва ли такой подход можно считать оправданным: клиническая картина, темпы протекания, прогноз, стратегия симптоматического лечения – все это детерминируется рядом значимых факторов (прежде всего, преимущественной локализацией процесса) и отличается в достаточной степени, чтобы говорить именно о самостоятельных заболеваниях.
Вышесказанное в полной мере относится к спиноцеребеллярной атаксии. Это наследственное нейродегенеративное заболевание с выраженной собственной спецификой, которое может манифестировать в любом возрасте (обычно в интервале 5-40 лет) и отличается многообразием форм: к настоящему времени выделено и описано свыше двадцати сравнительно самостоятельных типов (SCA, SCA 2, болезнь Фридрейха и мн.др.).
Обязательно для ознакомления! Помощь в лечении и госпитализации!
Лечение в Италии
Русский Медицинский Сервер / Лечение в Италии / Центр лечения редких заболеваний в Милане / Спиноцеребеллярная атаксия — лечение в Италии
Спиноцеребеллярная атаксия включает в себя различные активно развивающиеся наследственные формы патологий, характеризующиеся нарушением координации движений. При таких поражениях организма человека основные изменения происходят в мозжечке, в спинном мозге, а также в стволе головного мозга. Среди всех известных наследственных болезней спиноцеребеллярные атаксии находятся на втором месте по частоте возникновения после нервно-мышечных патологий.
Заболевание отличается заметным клиническим многообразием симптомов, есть широкая градация между смешанными формами патологии и мозжечковыми типами. Известно множество различных классификаций, основанных на клинико-анатомическом принципе и на типах генетического наследования признаков заболевания.
Важно знать, что все известные классификации спиноцеребеллярных дегенеративных процессов не в состоянии полноценно удовлетворить требования медицины. В основном это можно объяснить недостаточным изучением болезни и отсутствием основных представлений о развитии биохимических дефектов, которые становятся причинными.
Спиноцеребральная атаксия встречается с частотой в разных популяциях приблизительно в 1 — 23 случаях на сто тысяч населения. Патология обладает неравномерным этническим и географическим распространением. В России и в Италии в основном преобладает разновидность первого типа болезни, в Индии — чаще встречается второй тип спиноцеребеллярной атаксии, заболевание третьего типа чаще всего диагностируется у населения Германии, США и Японии.
Группа спиноцеребеллярных атаксий относится к повреждениям мозжечка, которые наследуются по аутосомно-доминантному типу. При этом происходящие в генах мутации провоцируют формирование патологических белковых продуктов, вызывающих отмирание и гибель клеток в мозжечке, спинном мозге, в коре полушарий головного мозга.
Неврологические симптомы спиноцеребеллярной атаксии развиваются довольно медленно, а само по себе развитие может затягиваться на срок до двадцати лет, но может встречать и более быстрое прогрессирование заболевания. Порой наблюдаются периоды стабильного состояния.
При развитии сопутствующих инфекционных поражений начинают проявляться дополнительные симптомы. Пациенты с сильно запущенными стадиями патологии лишаются возможности вставать с постели, у них формируется дисфагия, то есть нарушение акта глотания, и другие подобные признаки. Человек может умереть в связи с истощением и часто от развития миокардита, сопровождающегося тяжелыми формами недостаточности сердца. При надлежащем уходе больной доживает до сорока — пятидесяти лет.
Впоследствии симптоматика дополняется тремором ног и рук при реализации каких-либо действий и при нарушении координации движений. Кроме того, начинается изменение почерка — он становится неровным, а буквы слишком большими, также изменяется речь.
Для заболевания характерны и расстройства движения глаз — толчкообразные, резкие движения глазных яблок при смещении взгляда с одного объекта на другой. Часто отмечается возникновение нарушений глотания, произношения речи, нарушения работы слухового аппарата, нарушение стула и отхождения мочи, паралич ног и рук, развиваются патологические рефлексы. А привычные рефлексы одновременно ухудшают реакции и затем исчезают полностью.
Временами заболевания протекает в абортивной или легкой форме и провоцирует незначительную инвалидность пациента или вовсе не сопровождается инвалидностью. Такие патологии могут диагностироваться у родственников больного, которые страдают развернутыми клиническими типами болезни.
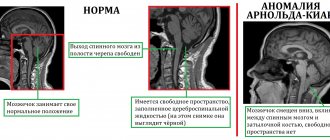
При диагностике предпочтение отдается методам визуализации, таким, как компьютерная или магнитно-резонансная томография. Они позволяют определить участки дегенерации нервных волокон, демиелинизацию нейронов моста, расширение всех компонентов системы циркуляции ликвора в головном мозгу, атрофические изменения коры полушарий головного мозга.
Для постановки диагноза также необходимо исключить и некоторые другие заболевания, протекающие со сходной клиникой (такие, как опухоли задней черепной ямки, рассеянный склероз, гидроцефалией, сосудистой патологией головного мозга, особенно в позвоночно-базиллярном отделе. Если на основании клиники удается подтвердить развитие атаксии, то гораздо более сложным является определение типа заболевания (если нет строго специфичных симптомов).
В настоящее время существует несколько мнений относительно тактики лечения данной патологии. Сторонники первой придерживаются мнения, что атаксия не подвержена медикаментозному лечению. В таком случае, лучше всего проводить поддерживающую терапию, способствующую замедлению прогрессирования заболевания. В обязательный комплекс входят ЛФК (вестибулярные тренировки), а также многие методы социальной, бытовой и трудовой реабилитации.
Другая же теория предполагает использование некоторых препаратов, однако, лечение проводится сугубо симптоматическое или стимулирующее. Назначаются инъекции витаминов, а также их пероральный прием. Как элемент такой терапии, возможно назначение противосудорожных препаратов (по показаниям). Лечение спиноцеребеллярной атаксии поддерживается и некоторыми физиотерапевтическими процедурами (например, электростимуляция).
! Несмотря на то, что многие из описанных в данном разделе болезней считаются неизлечимыми, в Центре лечения редких заболеваний в Милане постоянно ведется поиск новых методов. Благодаря генной терапии удалось добиться выдающихся результатов и полностью излечить некоторые редкие синдромы.
Обратитесь к консультанту на сайте или оставьте заявку — так вы можете узнать, какие методы предлагают итальянские врачи. Возможно, данное заболевание уже научились лечить в Милане.
+7 (925) 50 254 50 – срочное лечение в Италии
ЗАПРОС в КЛИНИКУ
2.Причины
В основе спиноцеребеллярных атаксий лежит наследуемая мутация определенных генов (тип наследования, как правило, таков, что если оба родителя являются носителями, то вероятность «срабатывания» патологии у ребенка составляет 1/4 или 25%). В результате нарушается ряд сложнейших электрохимических процессов, управляющих энерго- и белковым балансом (значительную роль играет дефектная структура белка фратаксина), передачей нервных импульсов от центра к периферии и обратно, и пр. Объединяющей особенностью для всей группы атаксий является то, что в нейронную дегенерацию вовлекаются структуры как головного мозга (прежде всего, мозжечок), так и спинного, а также проводниковые пути между ними, периферические нервы и, в отдельных случаях, ткань миокарда. Термин «атаксия» в дословном переводе означает «отсутствие порядка, согласованности», и, по определению, главным проявлением спиноцеребеллярной атаксии становятся прогрессирующие нарушения координации движений и, вообще, нервно-мышечной согласованности.
Посетите нашу страницу Неврология
Спиноцеребеллярные атаксии
Несмотря на значительное генетическое и отчасти клиническое разнообразие спиноцеребеллярных атаксий, молекулярные механизмы генетических нарушений при этих заболеваниях очень сходны. Основная причина патологии заключается в изменении количества тринуклеотидных последовательностей (CAG) в кодирующей части ассоциированных с заболеванием генов. Это приводит к увеличению количества аминокислоты глутамина в полученном белке, что изменяет физико-химические свойства протеина и нарушает его функции. В ряде случаев вышеуказанные белки прямо или косвенно участвуют в метаболизме нервной ткани, поэтому изменение их структуры приводит к спиноцеребеллярной атаксии. В настоящее время лучше всего изучены молекулярные механизмы 6 основных разновидностей этого заболевания – данные формы патологии встречаются наиболее часто и в совокупности составляют более 90% случаев спиноцеребеллярной атаксии.
Спиноцеребеллярная атаксия 1-го типа
считается самым распространенным и самым изученным вариантом данной патологии. Ее причиной выступают мутации в гене ATXN1, который располагается на 6-й хромосоме. В норме данный ген имеет не более 36 CAG-повторов, увеличение их количества приводит к развитию заболевания. Продуктом экспрессии гена ATXN1 является особый ДНК-связывающий белок, активно участвующий в метаболизме клеток Пуркинье мозжечка – при наличии мутантной разновидности гена это приводит к появлению агрегантов и постепенной дегенерации, что и становится причиной спиноцеребеллярной атаксии.
Спиноцеребеллярная атаксия 2-го типа
– менее распространенный вариант заболевания, этиология не так тщательно изучена. Причиной патологии является увеличение количества CAG-повторов в гене ATXN2, локализованном на 12-й хромосоме. В здоровом варианте гена количество вышеуказанных последовательностей составляет от 15 до 36, тогда как при спиноцеребеллярной атаксии их может быть свыше 100. Функции белка, который кодируется геном ATXN2, на сегодняшний момент неизвестны.
Спиноцеребеллярная атаксия тип 3
(другое название – болезнь Мачадо-Джозефа в честь двух больных, у которых впервые было описано данное состояние) – причиной этого варианта патологии выступают нарушения в гене ATXN3, расположенном на 14-й хромосоме. В норме количество CAG-повторов в этом гене не превышает 47, при развитии заболевания обнаруживается от 53 до 68 повторов. Данный ген кодирует белок, который предположительно участвует в энергетическом обмене нейронов мозжечка и базальных ядер.
Спиноцеребеллярная атаксия тип 6
– сравнительно редкий вид заболевания, обусловленный дефектами в гене CACNA1A, локализованном на 19-й хромосоме. Для развития патологии достаточно очень незначительного увеличения количества CAG-повторов – если в нормальном варианте гена их обнаруживают 5-20, то при наличии атаксии – 21-26. Ген CACNA1A кодирует белок-субъединицу кальциевых каналов, расположенных на нейронах мозжечка. Помимо спиноцеребеллярной атаксии, нарушения в гене CACNA1A обуславливают развитие эпизодической атаксии и некоторые наследственные формы мигрени.
Спиноцеребеллярная атаксия тип 7
– данная разновидность патологии вызывается нарушениями структуры гена ATXN7, который располагается на 3-й хромосоме. У здорового человека количество CAG-повторов составляет не более 35, тогда как при заболевании их количество может достигать нескольких сотен. Функции белка, который кодирует ген ATXN7, на сегодняшний момент изучаются.
Спиноцеребеллярная атаксия тип 8
обусловлена генетическим дефектом гена ATXN8, расположенного на 13-й хромосоме. Как и в других случаях, суть генетического дефекта при этом состоянии заключается в изменении количества тринуклеотидных последовательностей CAG – обычно их около 15-50, тогда как при патологии количество повторов может составлять свыше 1200.
Практически при любом типе спиноцеребеллярной атаксии патологическая форма белка, чрезмерно богатая глутамином, формирует отложения в ядрах или цитоплазме нейронов мозжечка и базальных ядер в виде плотных агрегатов. Этот процесс идет тем быстрее, чем сильнее количество CAG-повторов в ключевом гене отличается от нормы. Этим же объясняется механизм антиципации симптомов спиноцеребеллярной атаксии – в процессе мейоза при образовании половых клеток количество вышеуказанных тринуклеотидных последовательностей может увеличиваться, что приводит к усилению симптомов.
Так как подобное явление чаще имеет место при формировании мужских половых клеток, это становится причиной так называемой «отцовской передачи», когда антиципация регистрируется только при передаче заболевания от отца потомству. Многие врачи-генетики полагают, что основная причина спиноцеребеллярных атаксий лежит не в увеличении «гистидиновых» тринуклеотидов, а в делеции так называемых регулирующих триплетов, разделяющих участки CAG-повторов. Например, при первом типе заболевания это CAT, при втором CAA – они регулируют количество CAG-повторов и сохраняют стабильность их количества во время мейоза.
3.Симптоматика, диагностика
Вероятные симптомы спиноцеребеллярной нейронной дегенерации настолько полиморфны, что описать хотя бы основные из двадцати ее типов в одной статье нет никакой возможности. Отмечаются расстройства зрительно-моторной координации и мышечного тонуса (тремор, экстрапирамидная «скованность»); частичные параличи глазодвигательных мышц и дегенеративные ретинопатии в сочетании с атрофией зрительного нерва; общая атрофия мышечных волокон; разнообразные неврологические симптомокомплексы. Если нейродегенеративный процесс затрагивает кору головного мозга, постепенно развивается деменция – ослабоумливающее снижение когнитивных функций (память, внимание, различные виды распознавания и пр.), логического мышления, организации речи. В случае поражения продолговатого мозга развивается «бульбарная» симптоматика: деградация глотательного, небного, жевательного, дыхательного рефлексов.
Заболевания этой группы прогрессируют относительно медленно: течение может занимать до 20 лет и более, хотя описаны и значительно более быстрые развития. Больные постепенно утрачивают способность к самообслуживанию и, вообще, к продуктивному контакту с миром; они все больше зависят от опеки и ухода со стороны окружающих, к терминальной стадии впадая в полную беспомощность и погибая, как правило, от присоединившихся пневмоний, истощения, дыхательной недостаточности и пр.
Нейродегенеративные заболевания, в том числе атаксии, диагностируются клинически, в ходе неврологического осмотра и тщательного анализа жалоб и анамнестических сведений. Дополнительно могут назначаться методы томографической визуализации, нейропсихологическое обследование и пр., однако окончательно диагноз устанавливается и дифференцируется, как правило, лишь патоморфологически.
О нашей клинике м. Чистые пруды Страница Мединтерком!
Спиноцеребеллярная атаксия 1 типа (SCA 1)- тяжелое нейродегенеративное прогрессирующее заболевание с поздним возрастом манифестации, наследуется по аутосомно-доминантному типу; клинически характеризуется сочетанием нарастающих расстройств координации движений с признаками мультисистемного поражения головного и спинного мозга (Иллариошкин и др., 2002). Начало симптомов при СЦА1 — наблюдается обычно во взрослой жизни, в среднем — в 30 лет. Если симптомы появляются раньше — до 20 лет, дополнительно к атаксии часто встречаются и другие симптомы. В случаях очень раннего (до 13 лет) болезнь может быть более тяжелой и иметь быстрое прогрессирование.
| Основные клинические симптомы: первым симптомом обычно является нарушение координации рук и нарушение баланса при ходьбе. Фактически, само слово «атаксия» означает нарушение координации. При прогрессировании СЦА1 в течение нескольких лет появляются трудности при глотании и неясная речь. В некоторых случаях, у больных появляются дополнительные признаки типа нейропатии (потери чувств и рефлексов в ногах), мышечной спастичности, слабости, или потери памяти. |
При СЦА1 генетические дефекты ведут к ухудшению работы определенных нервных волокон, несущих информацию к головному мозгу и от него, которая заканчивается дегенерацией мозжечка (координационного центра мозга). Часто отмечается кровное родство родителей. Заболевание не поддается лечению, смерть чаще всего наступает спустя 10-15 лет от момента появления первых симптомов. Генетическая причина развития СЦА1 заключается в экспансии числа копий тандемных тринуклеотидных CAG-повторов в кодирующей области гена SCA1 (Orr et al., 1993). Для СЦА1 возможна прямая ДНК-диагностика заболевания на пресимптомной стадии, иногда за много лет до появления каких-либо неврологических и/или психических расстройств. Это имеет особое значение для медико-генетического консультирования, поскольку в настоящее время не найдены еще эффективные способы лечения СЦА1, хотя исследования в этом направлении активно ведутся в мире (Ogawa, 2004; Dueñas et al., 2006; Takei et al., 2007; Gao et al., 2008). Единственным способом борьбы с этим заболеванием на сегодняшний день является профилактика появления новых случаев СЦА1 в отягощенных семьях (Иллариошкин и др., 1997). До середины 70-х годов в Якутии болезнь относили к «мозжечковой форме» вилюйского энцефаломиелита. В настоящее время спиноцеребеллярная атаксия I типа рассматривается как этноспецифическое наследственное заболевание (Пузырев, Максимова, 2008).
Высокая распространенность заболевания (38,6 на 100 тыс. якутов по сравнению 1-2:100 тыс. в мировом населении) в Якутии была оценена как «сибирский очаг» накопления заболевания, крупнейший в мире. Районы высокого накопления СЦА1 — Абыйский и Усть-Алданский улусы Якутии — характеризуются однородным национальным составом; высоким уровнем рождаемости; низким уровнем миграций. Молекулярной основой заболевания является увеличение у якутов числа тринуклеотидных CAG-повторов до 39-71 по сравнению с 19-36 в норме в гене SCA1, локализованного в области 6p22 – p23. Среди различных форма наследственных атаксий в Якутии СЦА 1 встречается в 88,1% всех семей.
Литература:
- Кучер А.Н., Данилова A.JL, Конева Л.A., Максимова Н.Р., Ноговицина А.Н. Генетико-демографическое изучение народонаселения Республики Саха (Якутия) // Якутский медицинский журнал. — 2005. № 2(10). — С. 4-12.
- Конева Л. A., Кучер А.Н., Максимова Н.Р., Пузырёв В.П. Подходы к моделированию распространенности спиноцеребеллярной атаксии I типа в изолированной популяции // Генетика человека и патология: Сб. науч. трудов / Под ред. В.П. Пузырева. — Вып. 7. — Томск: Печатная мануфактура, 2004. -С. 92-101.
- Конева Л .А., Кучер А.Н., Пузырев В.П., Ноговицына А.Н., Максимова Н.Р., Сухомясова А.Л., Данилова А.Л. Демографические и клинико-генетические особенности распространенности спиноцеребеллярной атаксии I типа в Усть-Алданском и Абыйском улусах республики Саха (Якутия) / Матер, на-уч.-практич. конф. «Актуальные вопросы профилактической медицины». -Улан-Удэ. — 2005. — С. 97-100.
- Конева Л.А., Кучер А.Н., Пузырёв В.П., Максимова Н.Р., Ноговицина А.Н., Сухомясова А.Л., Данилова А.Л., Платонов Ф.А., Коротов М.Н. Характеристика заболеваемости спиноцеребеллярной атаксией I типа в Усть-Алданском и Абыйском улусах Республики Саха (Якутия) / Матер. Между-нар. науч.-практич. конф. «Генетические аспекты патологии человека. Проблемы сохранения генофонда коренных народов Севера», Якутск: НИПК «Сахаполиграфиздат». — 2005. — С. 99-100.
- Конева Л.А., Кучер А.Н., Пузырёв В.П., Ноговицина А.Н., Максимова Н.Р., Сухомясова А.Л., Данилова А.Л. Распространенность спиноцеребеллярной атаксии I типа в Усть-Алданском и Абыйском улусах Республики Саха (Якутия) / Матер. Итоговой науч.-практ. конф. ГУ НИИ медицинских проблем севера СО РАМН «Вопросы сохранения и развития здоровья населения Севера и Сибири», Красноярск. — 2005. — С. 127-129.
- Конева Л.А., Максимова Н.Р. Распространенность спиноцеребеллярной атаксии I типа в Якутии: проблемы и пути решения / Матер, конкурса работ молодых ученых «Теоретические и прикладные проблемы медицинской генетики» СО РАМН, Новосибирск. — 2004. — С.103-109.
- Конева Л .А., Максимова Н.Р., Кучер А.Н., Пузырёв В.П. Динамика частоты спиноцеребеллярной атаксии I типа у якутов с учетом специфики популяционной структуры / Генетика в XXI веке: современное состояние и перспективы развития: Матер. 3-го Съезда ВОГиС. -М. — 2004. — С.28.
4.Лечение
В настоящее время не существует лечения, которое обращало бы вспять или хотя бы останавливало процессы нейродегенерации. Все виды практикуемой сегодня терапии носят сугубо паллиативный характер и направляются на смягчение наиболее дезадаптирующих, снижающих качество жизни симптомов, доминирующих в конкретной клинической картине. Как правило, назначают препараты для улучшения нейротрофики (питания нервных тканей), витаминные комплексы, массаж, лечебная физкультура для коррекции и/или компенсации двигательных расстройств.
Описание
Синонимы (rus): Болезнь Мачадо-Джозефа
Синонимы (eng): Spinocerebellar ataxia type 3, SCA3
Биоматериал: Венозная кровь
Показатель(и): Экспансия в гене ATXN3
Метод(и): Полимеразная цепная реакция (ПЦР)
Тип контейнера и особенности преаналитики: Пробирка для гематологических исследований с EDTA, 2 мл (фиолетовая крышечка)
Спиноцеребеллярная атаксия 3 типа (СЦА 3) – представляет собой нейродегенеративное прогрессирующее генетическое заболевание. В основе патогенеза лежит экспансия CAG-тринуклеотидных повторов нестабильного участка гена ATXN3. При количестве менее 60 CAG-триплетных повторов заболевание исключается. Заболевание наследуется по аутосомно-доминантному типу, характерен феномен антиципации, количество повторов обратно пропорционально коррелирует со временем манифестации заболевания и прямо пропорционально с тяжестью его течения. Заболевание очень гетерогенно и может проявляться 5-ю вариантами: 1 тип – ригидность, спастичность, брадикинезия без атаксии; 2 тип – атаксия, поражение верхних моторных нейронов; 3 тип – атаксия, полинейропатия; 4 тип – L-ДОПА зависимый паркинсонизм; 5 тип – спастическая параплегия. Исследование рекомендуется пациентам с поражением верхних моторных нейронов, атаксией, полинейропатией.