General information
A characteristic feature of the disease is metabolic disorders in skeletal muscle tissue. The muscles of a sick child lose their function partially or entirely, that is, weakness appears in them and the range of movements decreases. The quality of life is significantly reduced. Source: Komantsev V.N., Skripchenko N.V., Sosina E.S., Klimkin A.V. POLYNEUROPATHY AND MYOPATHY OF CRITICAL CONDITIONS IN ADULTS AND CHILDREN: DIAGNOSIS, CLINICAL MANIFESTATIONS, PROGNOSIS, TREATMENT // Modern problems of science and education. – 2012. – No. 5
This pathology usually has a hereditary form and can be diagnosed in children of any age. It is not life-threatening, except in cases where atrophy of the heart muscle and respiratory muscles occurs. Source: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2796972/ Chris M. Jay, Nick Levonyak, Gregory Nemunaitis, Phillip B. Maples and John Nemunaitis Hereditary Inclusion Body Myopathy (HIBM2) Gene Regul Syst Bio. 2009; 3: 181–190.
The disease has a number of complications:
- development of respiratory failure;
- limited mobility;
- paralysis;
- congestive pneumonia;
- depressive, suicidal mood of the patient;
- increased risk of death.
Duchenne muscular dystrophy: issues for therapy with antisense oligonucleotides
"Amondis 45" - by analogy with "Exondis 51", "Viondis 53" and "Viltepso" - received regulatory approval conditionally: on the basis of a surrogate endpoint. These four drugs produced statistically significant increases in dystrophin protein levels in skeletal muscle, which are reasonably predictive of clinical benefit for patients. The FDA issued a positive verdict due to the life-threatening and disabling nature of Duchenne muscular dystrophy, for which reliable and highly effective specific therapy is still not proposed.
According to industry experts, the clinically significant effectiveness of any therapy for Duchenne muscular dystrophy is the achievement of de novo synthesis of dystrophin to at least 10% of its normal production in a healthy body. In fact, Exondis 51, which raised dystrophin levels to just 0.44% after a 48-week course of treatment, was called by some critics a “scientifically elegant placebo.”
By the way, the European Medicines Agency (EMA) refused to approve Exondis 51, citing a lack of adequate clinical data to confirm that the modest increase in dystrophin levels provided by eteplirsen would bring long-term benefits to patients. Perhaps the regulator will change its decision when the results of the 144-week phase III clinical trial MIS51ON (NCT03992430), testing Exondis 51 at standard and up to 6 times higher doses, are collected by 2026.
The ongoing ESSENCE clinical trial (NCT02500381), which will be completed by 2024, is designed to confirm the therapeutic efficacy of Amondis 45 and Viondis 53. After 96 and 144 weeks of treatment, patients' muscle functionality will be assessed: 6MWT test, ability to rise independently from the floor, time to loss of ambulatory status, North Star Ambulatory Assessment (NSAA), FVC score. In addition, after 96 weeks of therapy, the level of dystrophin in muscle biopsies will be measured (by Western blotting and immunohistochemistry).
Causes of myopathy in children:
- hormonal imbalances;
- heredity;
- genetic defects (deficiency of an enzyme that ensures metabolic processes in muscles; a defect in a cell that plays the most important role in the delivery of energy material to the muscles); Source: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5575512/ Alessia Nasca, Chiara Scotton, Irina Zaharieva, Marcella Neri, Rita Selvatici, Olafur Thor Magnusson, Aniko Gal, David Weaver, Rachele Rossi, Annarita Armaroli, Marika Pane, Rahul Phadke, Anna Sarkozy, Francesco Muntoni, Imelda Hughes, Antonella Cecconi, György Hajnóczky, Alice Donati, Eugenio Mercuri, Massimo Zeviani Recessive mutations in MSTO1 cause mitochondrial dynamics impairment, leading to myopathy and ataxia Hum Mutat. 2021 Aug; 38(8): 970–977.
- systemic connective tissue lesions.
Causes of muscular dystrophy
Muscular dystrophy is a muscle disease, or more precisely, a group of diseases that leads to weakness and degenerative changes in the muscles.
Dystrophy most often affects skeletal muscles. They gradually weaken, lose their ability to contract, decrease in volume and are replaced by connective or fatty tissue. The cause of the disease is a mutation of the gene that is responsible for the synthesis of proteins in muscle cells. Various types of genes can be affected, both those located on the sex chromosome and those not associated with it. The mechanism of inherited transmission of the disease depends on this (for example, the Duchenne gene, associated with the sex X chromosome, is transmitted through the male line; women do not suffer from dystrophy caused by this gene, although they are its carriers).
Symptoms and treatment of pathology in a child
Clinical signs of myopathy in children:
- change in gait;
- weakness that does not go away with rest;
- delayed motor development;
- flaccid, flabby muscles;
- atrophy (thinning) of muscles;
- curvature of the spine is a manifestation indicating weakness of the muscular corset.
Negative processes manifest themselves in children in early and teenage years, but since myopathy develops slowly, it can go undetected for a long time. In addition, children are able to compensate for muscle deficiency by using other, healthy muscles more actively.
The most common changes are observed in the areas of the shoulders, legs, arms, pelvis, and chest. With this disease they are always bilateral and symmetrical.
As the disease progresses, movement disorders appear:
- it is difficult for a child to sit up from a lying position;
- movements are abnormal, “wrong”;
- when walking and/or running, fatigue quickly sets in;
- the child has difficulty maintaining balance and often falls;
- It is difficult for a child to climb stairs.
Disturbances in appearance may also appear:
- protruding ribs;
- very thin, as if overtightened, waist;
- flattened chest;
- slouch;
- Irregular shape of legs - thick calves and thin thighs.
"Amondis 45": safety of casimersen in the treatment of Duchenne muscular dystrophy
Studies in male mice and rats have demonstrated renal toxicity of casimersen, manifested by renal tubular degeneration. Although human trials have not reported anything similar, it is important to consider that some other antisense oligonucleotides have been studied in patients who have experienced renal toxicity, including potentially fatal glomerulonephritis. Therefore, therapy with Amondis 45 should be accompanied by monitoring of renal function.
The most common adverse reactions to casimersen were: upper respiratory tract infections (65% of patients versus 55% in the placebo group), cough (33% versus 26%), fever (33% versus 23%), headache (32). % vs. 19%), joint pain (21% vs. 10%), pain in the oropharynx (21% vs. 7%).
Diagnosis of myopathy
The disease is expressed:
- increasing symptoms;
- absence of seizures and neurological manifestations;
- selective localization;
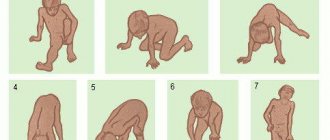
- characteristic "duck" gait.
For an accurate diagnosis, first of all, an anamnesis is collected to find out whether there have been cases of this disease in the family. Then an examination is carried out by a neurologist, during which the doctor evaluates muscle tone, the spread of weakness, the presence of muscle thinning, the degree of body deformation, the severity of reflexes, gait, and asks the child to sit up from a lying position and stand up from a sitting position.
Laboratory diagnostics include:
- clinical blood test;
- muscle biopsy;
- checking thyroid hormone levels.
A genetic examination of the child and close relatives is also carried out. Source: https://www.mda.org/disease/congenital-myopathies/diagnosis The Muscular Dystrophy Association (MDA).
Diagnostics
Genetic analysis is considered an extremely informative method for diagnosing SMA. It can be performed on children and adults at any age, and for the purpose of early diagnosis, it can be performed even at the stage of intrauterine development. If DNA analysis is not possible and for final confirmation of the diagnosis, the following are prescribed:
- blood chemistry;
- histological analysis of muscle fibers;
- MRI;
- electromyography;
- spinal cord microscopy;
- tandem mass spectrometry.
You should not postpone the operation until later, as progressive changes can lead to severe breathing problems. This will be an absolute contraindication to surgical treatment. Medical surgeons carry out preparatory actions, the operation itself and form a rehabilitation recovery system at the level of European clinics at an affordable cost.
Types of disease
One of the classification features is the cause of the disease . According to it, myopathy is distinguished:
- primary (appears independently at birth, in early childhood or adolescence);
- secondary (develops against the background of other diseases).
According to the location of weakness, the disease is:
- proximal (muscles are weakened closer to the body);
- distal (muscles are weakened in the limbs further from the body);
- mixed.
The following forms of the disease also exist :
- Pseudohypertrophic (Duchenne-Griesinger). Appears at 3-6 years of age, rarely before one year. Mainly affects the muscles of the legs and pelvis. Associated lesions: weakness of the respiratory and cardiac muscles. There is a high probability of death even before adulthood.
- Landouzy-Dejerine. Begins at 10-15 years of age and affects the face. The facial muscles weaken, the lips protrude and thicken, and often the patient cannot close his eyelids. Then the muscles are involved in a descending manner down to the shoulder girdle.
- Erba-Rotta (youth). The onset of the disease is 10-20 years, boys are mainly susceptible to this form. The processes take place from top to bottom or bottom to top, rarely throughout the entire body or in the face area.
Important!
Congenital myopathy is one of the most dangerous forms in children, often resulting in death. Her treatment is limited to improving vitality and begins in the first months after birth. The main thing in therapy is the prevention of respiratory failure, the organization of tube feeding. As the child grows, orthopedic correction techniques are used, physiotherapy and social adaptation are of great importance.
Congenital myopathy
Central core disease is inherited in an autosomal dominant manner; isolated sporadic cases of the disease are also known. Congenital myopathy of this type is manifested by delayed motor development during the first year of life, is less often found in adult patients, and is often accompanied by weakness of facial muscles. The patients are characterized by their small stature and fragile figure, and the presence of skeletal deformities. Patients with this type of congenital myopathy have an increased risk of malignant hyperthermia. A biopsy of muscle tissue reveals muscle fibers with single or multiple zones of aseptic necrosis.
Nemaline congenital myopathy includes actinopathy, nebulinopathy, tropomyosinopathy and troponinopathy. Its inheritance occurs most often according to an autosomal dominant principle, but recessive inheritance and sporadic cases of incidence also occur. The classic form of nemaline congenital myopathy is characterized by floppy baby syndrome. The severe form manifests itself in the prenatal period in the form of fetal akinesia, and at the birth of a child - severe motor impairment, weakness of the facial muscles and respiratory failure. A mild form of this type of congenital myopathy is diagnosed after early childhood, sometimes during adolescence, and occurs without weakness of the facial muscles. There is also a specific form of nemaline congenital myopathy, in which the development of ophthalmoplegia, cardiomyopathy, and rigid spine syndrome is possible. Morphological examination reveals the presence of characteristic rod- or thread-like bodies in the muscles.
Myotubular congenital myopathy is most often inherited as an autosomal one, in which muscle weakness is mild and can be observed in both girls and boys. X-linked myotubular congenital myopathy affects only males and is characterized by a more severe course with weakness of the facial muscles, swallowing and respiratory problems. In the muscle tissue biopsy, damage to type I fibers predominates. The central location of the myocyte nuclei is noted, which corresponds to the muscle tissue of the embryo at 8-10 weeks of pregnancy. In this regard, most researchers consider myotubular myopathy as a result of underdevelopment of muscle tissue.
Multiple core myopathy is most often observed as an autosomal recessive disease, although a dominant mode of inheritance is also possible. Muscle weakness in the proximal parts is typical, which is observed in infancy. Much less often, the disease debuts at an older age. In such cases, generalized muscle weakness is noted. In a muscle biopsy, cells with the absence of mitochondria, destruction of sacromeres and wasting of muscle fibers are determined.
Congenital disproportion of muscle fiber types is manifested by generalized muscle weakness, including facial muscles, muscle hypotonia, and skeletal abnormalities. The mode of inheritance of this congenital myopathy has not yet been established. In muscle biopsy, an increase in the number and small size of type I fibers is observed against the background of hypertrophy or normal size of type II fibers.
Treatment methods
Important!
The sooner you start treating a child, the greater his chances of a fairly high quality of life.
Treatment boils down to the following activities:
- injection of adenosine triphosphoric acid (ATP) in courses;
- iontophoresis;
- vitaminization;
- drugs to improve blood circulation;
- massage;
- use of orthopedic correction devices by patients;
- the use of drugs for better neuromuscular conduction;
- hormone therapy;
- and etc.
The hereditary form of the disease cannot be completely cured, but it is possible to specifically eliminate the main symptoms by:
- orthopedic correction;
- regular and breathing exercises.
Sometimes surgery is required. It is aimed at correcting scoliosis that occurs against the background of the underlying disease.
Promising methods for treating myopathy are: the use of stem cells and gene therapy.
Gene therapy for Duchenne muscular dystrophy: long hope
Sarepta continues to develop gene therapy for Duchenne muscular dystrophy, which seems to be suitable for all patients, regardless of the mutational background of the disease. There is hope that after a single injection, the course of this disabling and fatal disease will change dramatically: patients will not only get rid of the threat of imminent death, but will also get a chance for a more or less full independent life.
Sarepta's efforts are focused on virus-vector delivery of a synthetic transgene encoding the most important domains of the dystrophin protein, the critical deficiency of which is the cause of Duchenne muscular dystrophy. Since the adenoviral vector has a limited ability to transfer genetic information (the capacity is approximately 4.7 kilobases), it is not able to transport the dystrophin gene, which is too large (2.4 megabases), so a truncated but still functional version of the latter will do just fine - so called microdystrophin.
In early January 2021, Sarepta shared key results from the ongoing phase II (randomized, double-blind, placebo-controlled, multicenter) clinical trial NCT03769116 of the gene therapy program SRP-9001 (rAAVrh74.MHCK7.micro-dystrophin), including boys (n=41 ) aged 4–7 years with Duchenne muscular dystrophy.
After 12 weeks after a single dose of delandistrogene moxeparvovec (SRP-9001), the level of microdystrophin expression reached 28.1% (p<0.0001). Proper biological indicators were also demonstrated: the number of copies of virus-vector genomes per nucleus (1.56), the proportion (33.0%) and staining intensity (63.7%) of dystrophin-positive muscle fibers, and a decrease in creatine kinase.
After 48 weeks, the initial total NSAA score in the gene therapy group increased by 1.7 points (p=0.0090) versus an increase of 0.9 points (p=0.1411) in the control group. However, the difference was not statistically significant (p=0.37).
The 17-item NSAA rating scale is used to measure functional motor skills in children with Duchenne muscular dystrophy and is used to monitor disease progression and therapeutic effects. The NSAA scale takes into account the patients' ability to stand, walk, get up from a chair and the floor, stand on one leg, go up and down stairs, jump, run, etc.
Against the backdrop of such disappointing news, Sarepta’s stock quotes immediately lost half their value: minus $7 billion of the company’s market value.
“Sarepha,” however, is not upset, explaining the formal failure of the test as follows. The randomization of participants was stratified by age groups: 4–5 and 6–7 years. In the first group, the total NSAA score among those who underwent gene therapy increased by 4.3 points (p<0.0001) - versus its increase of 1.9 points (p=0.0126) among those who received placebo. The 2.5-point difference was statistically significant (p=0.0172), which was said to be due to patients' baseline performance status, which was well balanced between the gene therapy and placebo groups. However, in the second group this was characterized by a clear discrepancy: the placebo group actually included patients with a less severe severity of Duchenne muscular dystrophy, when compared with patients in the experimental treatment group. In other words, the therapeutic failure of the delandistrogen moxeparvovec among the entire population of patients included in the study is explained not by the fact that the gene therapy being studied is ineffective, but by errors in the selection of subjects.
Be that as it may, the clinical trial continues.
At the end of December 2021, Sarepta granted Roche all commercial rights to SRP-9001 outside the United States. In exchange, it received an advance of $750 million in cash and $400 million in shares, with additional payments of up to $1.7 billion promised as it passed regulatory and sales milestones, plus royalties from sales.
Pfizer and Solid Biosciences are also involved in gene therapy for Duchenne muscular dystrophy, testing fordadistrogene movaparvovec (PF-06939926) and SGT-001, respectively.
Advantages of contacting SM-Clinic
Our clinic employs some of the best pediatric neurologists in St. Petersburg, doctors of high categories with impressive experience. Your child will be able to undergo diagnostics using modern equipment, undergo laboratory tests without queues and in comfortable conditions. SM-Clinic specialists will develop an optimal treatment plan in a short time, taking into account the individual characteristics of the patient and the form of his disease.
Call us for additional questions and to schedule an appointment.
Sources:
- Komantsev V.N., Skripchenko N.V., Sosina E.S., Klimkin A.V. Polyneuropathy and myopathy of critical conditions in adults and children: diagnosis, clinical manifestations, prognosis, treatment // Modern problems of science and education, 2012, No. 5.
- https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2796972/ Chris M. Jay, Nick Levonyak, Gregory Nemunaitis, Phillip B. Maples and John Nemunaitis Hereditary Inclusion Body Myopathy (HIBM2) Gene Regul Syst Bio. 2009; 3: 181–190.
- https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5575512/ Alessia Nasca, Chiara Scotton, Irina Zaharieva, Marcella Neri, Rita Selvatici, Olafur Thor Magnusson, Aniko Gal, David Weaver, Rachele Rossi, Annarita Armaroli , Marika Pane, Rahul Phadke, Anna Sarkozy, Francesco Muntoni, Imelda Hughes, Antonella Cecconi, György Hajnóczky, Alice Donati, Eugenio Mercuri, Massimo Zeviani
- Recessive mutations in MSTO1 cause mitochondrial dynamics impairment, leading to myopathy and ataxia Hum Mutat. 2021 Aug; 38(8): 970–977.
- https://www.mda.org/disease/congenital-myopathies/diagnosis The Muscular Dystrophy Association (MDA).
Pitsukha Svetlana Anatolyevna Clinic
Author of the article
Pitsukha Svetlana Anatolevna
Doctor of the highest qualification category
Specialty: neurologist
Experience: 24 years
The information in this article is provided for reference purposes and does not replace advice from a qualified professional. Don't self-medicate! At the first signs of illness, you should consult a doctor.
Signs
There are several stages of muscle wasting:
- The amount of subcutaneous tissue throughout the body decreases. A person loses up to 20% of body weight. He has pallor, weak appetite, muscle tone decreases;
- Subcutaneous tissue on the abdomen and chest practically disappears. The skin becomes gray, the muscles become flabby, and the liver increases in size. Mental health disorders and irritability occur;
- Severe wasting (cachexia) occurs when the patient loses 30% of muscle mass. The condition requires intensive therapy.
Common symptoms of muscle wasting include the following:
- Constant muscle pain;
- Weakness;
- Inability to perform normal movements;
- Significant loss of body weight.
If areas of muscle wasting are located symmetrically, this raises suspicion of myopathy or spinal amyotrophy. With progressive muscular dystrophy, relatively isolated wasting of the quadriceps femoris or biceps brachii is observed. If malnutrition is located in the distal limbs, we are talking about polyneuropathy (with impaired sensitivity and loss of reflexes in the distal limbs) or Steinert’s myotonic dystrophy.
Unilateral acquired isolated muscle wasting is always a consequence of damage to the root, plexus or peripheral nerve. Decisive for the topical diagnosis is the characteristic distribution of the process of malnutrition and sensory disturbances or prolonged inactivity of the muscle. Hypotrophy of the quadriceps muscle occurs with arthrosis of the knee joint and with sarcoma of the hip. Focal wasting of individual muscles or muscle groups, isolated and sometimes symmetrical, can slowly progress over many years. This is a sign of focal damage to the ganglion cells of the anterior horns or ischemia in the area of the blood supply of the artery.
Hypotrophy of the calf muscles often occurs. With progressive muscular dystrophy, sometimes in significantly hypotrophied muscles areas with intact muscle fibers are identified, which look like nodules. Doctors at the Yusupov Hospital distinguish them from a muscle roll, which is formed when the short head of the biceps brachii muscle is ruptured and is noticeable on the flexor surface of the shoulder.
Prices
| Name of service (price list incomplete) | Price |
| Appointment (examination, consultation) with a neurologist, primary, therapeutic and diagnostic, outpatient | 1750 rub. |
| Consultation (interpretation) with analyzes from third parties | 2250 rub. |
| Prescription of treatment regimen (for up to 1 month) | 1800 rub. |
| Prescription of treatment regimen (for a period of 1 month) | 2700 rub. |
| Consultation with a candidate of medical sciences | 2500 rub. |
| Transcranial duplex scanning (TCDS) of cerebral vessels | 3600 rub. |
Treatment
Neurologists at the Yusupov Hospital prescribe complex treatment to patients suffering from muscle dystrophy, aimed at eliminating the cause of the disease, influencing the mechanisms of development of the pathological process, and reducing the manifestations of the disease. To improve blood flow in peripheral vessels, angioprotectors (trental, pentoxifylline, chimes), low molecular weight dextran, and prostaglandin E preparations (vasaprostan) are used. After dilating blood vessels with no-shpa and papaverine, the supply of muscle fibers with oxygen and nutrients improves.
B vitamins (thiamine hydrochloride, pyridoxine hydrotartrate, cyanocobalamin) normalize metabolic processes and the conduction of nerve impulses. Biological stimulants stimulate the regeneration of muscle fibers and restore muscle volume: aloe, actovegin, plasmol. To restore muscle conduction, prozerin, galantamine, and armin are used.
Acquired muscle diseases
Acquired muscle diseases can be of inflammatory, infectious, drug, or toxic origin.
Inflammatory diseases of human muscle - polymyositis, dermatopolymyositis, inclusion body myositis, sarcoid myopathy.
Infectious myositis can be viral, bacterial, parasitic, etc. The main symptoms - muscle weakness and pain - appear acutely. Sometimes they may be accompanied by skin lesions (dermatopolymyositis). A distinctive feature of the course of these diseases is an increase in body temperature.