How to recognize SMA
Muscle atrophy in children diagnosed with SMA (spinal muscular atrophy) can begin at different times.
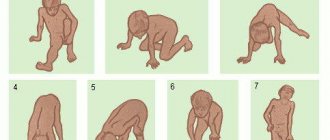
The most severe form of muscle atrophy in newborns appears in the first six months of life. The baby is lethargic and sucks poorly. At 3-4 months the child does not roll over on his own and does not attempt to crawl. With spinal muscular atrophy, the baby's posture resembles a “frog”. There is a type of disease that appears after 7-18 months. Muscle atrophy leads to regression of acquired skills in the child. The baby, who was crawling and starting to get up, suddenly becomes inactive. Over time, he stops sitting up straight. With spinal atrophy, reflexes from the upper and lower extremities disappear.
Symptoms of muscle atrophy with spinal muscular atrophy may appear closer to two years of age. Patients have already mastered the skills of standing and walking. At the same time, muscle atrophy confines them to a wheelchair. Intelligence, urination and defecation functions are preserved. SMA after 2 years is the mildest type of the disease.
Muscle atrophy (spinal muscular atrophy) has the following symptoms:
- Appears in infancy or childhood;
- Accompanied by disturbances in walking, running, and standing;
- Tremors and fasciculations (twitching) are detected;
- The “rollback” of motor skills is determined;
- With spinal atrophy, no impairment of intelligence or autonomic functions is detected.
If a child has these signs, he should be consulted with specialists. The diagnosis of SMA is made based on DNA testing.
IV type
The first signs and symptoms appear at 30-50 years of age. The main complaints are muscle weakness and tremors. Due to atrophy, contractures appear in the joint area, limiting their mobility. Patients lose weight sharply.
Often accompanied by curvature of the spine and deformation of the chest.
The muscles of the legs are the first to be involved in the process, then the arms are gradually involved. As a rule, respiratory and swallowing functions are not impaired.
Most patients walk independently but limp, experience pain and discomfort. In rare cases, the progression of amyotrophy leads to loss of the ability to walk, and patients are forced to move in a wheelchair.
WERDNIG-HOFFMANN SPINAL AMIOTROPHY
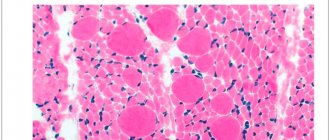
Inherited in an autosomal recessive manner. Underdevelopment of the cells of the anterior horns of the spinal cord, demyelination of the anterior roots, and similar changes in the motor nuclei and roots of the Y, YI, YII, IX, X, XI, XII cranial nerves are detected. In skeletal muscles, neurogenic changes are characterized by “bundle atrophy,” an alternation of atrophied and preserved bundles of muscle fibers.
CLINIC
There are three forms of the disease:
- congenital;
- early childhood;
- late childhood
.
In congenital form
children are born with flaccid paresis. From the first days of life, generalized muscle hypotonia and decreased or absent tendon reflexes are evident. Bulbar disorders are detected early, manifested by sluggish sucking, weak cry, tongue fibrillations, and decreased pharyngeal reflex. The disease is combined with osteoarticular deformities: scoliosis, funnel chest, joint contractures. The development of static and locomotor functions is sharply slowed down. Reduced intelligence. Congenital malformations are often observed: congenital hydrocephalus, cryptorchidism, hemangioma, hip dysplasia, clubfoot, etc.
The course is rapidly progressive and malignant. Death occurs before the age of 9 years. One of the main causes of death are severe somatic disorders (heart and respiratory failure), caused by weakness of the chest muscles and a decrease in its participation in the physiology of breathing.
With early childhood form
The first signs of the disease appear in the second half of life. The disease develops subacutely, often after infection or food intoxication. Flaccid paresis is initially localized in the legs, quickly spreading to the muscles of the trunk and arms. Diffuse muscle atrophy is combined with fasciculations, fibrillations of the tongue, fine tremor of the fingers, and tendon contractures. Muscle tone and tendon reflexes decrease. In the later stages, generalized muscle hypotonia and symptoms of bulbar palsy occur.
The course is malignant, death occurs by 14–15 years of age.
In late form
signs of the disease appear at 1.5 - 2.5 years. The disease begins unnoticed. Movements become awkward and uncertain. Children often trip and fall. The gait changes - they walk with their legs bent at the knees (the gait of a “wind-up doll”). Flaccid paresis is initially localized in the proximal muscle groups of the legs, then relatively slowly moves to the proximal muscle groups of the arms and trunk muscles; muscle atrophy is usually subtle due to the well-developed subcutaneous fat layer. Fasciculations, fibrillations of the tongue, fine tremor of the fingers, bulbar symptoms - fibrillations and atrophy of the tongue, decreased pharyngeal and palatal reflexes are typical. Tendon reflexes fade in the early stages of the disease. Osteoarticular deformities develop parallel to the underlying disease. The most pronounced deformation of the chest.
The course is malignant, but milder. Patients live up to 20 - 30 years.
Diagnostics.
Autosomal recessive type of inheritance, early onset, the presence of diffuse atrophies with predominant localization in the proximal muscle groups, generalized muscle hypotonia, fasciculations, fibrillations of the tongue, absence of pseudohypertrophies, progressive, malignant course, electromyography data and morphology of skeletal muscles, revealing the denervation nature of the changes.
Spinal muscular atrophy (amyotrophy) Werdnig-Hoffmann is a hereditary malignant disease, the onset of development of which occurs from birth to 1-1.5 years. This is one of the most severe forms of muscle atrophy. There is a diffuse increase in muscle atrophy throughout the body. The child loses the ability to sit and move independently, and paresis progresses.
The disease was first described by scientists Werdnig and Goffman. They proved the morphological essence of spinal amyotrophy. But they assumed the existence of only one form of the disease. Later, other scientists Welander and Kueckelberg described another form of spinal muscle atrophy. All variants of the disease have the same genetic nature. Today there are no methods that can completely cure this pathology. Therapeutic measures are aimed at improving the trophism of muscles and nervous tissue.
Treatment
The main goal of research aimed at treating spinal muscular amyotrophy is related to increasing the level of SMN protein. Currently, medications are being tested, and official Russian medicine does not use them.
Treatment today includes medications that improve the passage of nerve impulses. Nootropic drugs are prescribed, the main task of which is to improve brain function. Dietary supplements are prescribed to improve metabolism. Vitamin therapy is indicated, in particular, taking B vitamins.
Drugs affecting neuromuscular conduction:
- Alpha lipoic acid
- Acetyl L-carnitine
- Alpha glycerophosphocholine
Vitamins and vitamin complexes:
- Thiamine (B-1)
- Pyridoxine (B-6)
- B-complex
Important treatment methods are massage, physiotherapy, and neuromuscular stimulation. Exercise therapy is prescribed. Physical exercise helps maintain strength, on the other hand, doing it in society, going to the pool helps to socialize and communicate with other people.
Patients with SMA are advised to follow a diet. Food is a source of substances needed by muscles. Thus, the necessary amino acids are found in grains, meat, fish, mushrooms, nuts, and dairy products. Recommended dishes made from oats, wheat and brown rice.
We advise you to study - Symptoms, effective treatment of spinal osteoporosis, diagnosis of the disease
Spinach, broccoli, herring, onions, grapefruit, and watermelon will help to naturally maintain and grow muscles. To increase testosterone, men are recommended to take dill, parsnips, ginseng, and parsley.
Treatment
A practical patient's guide to the legal basis for the provision of medical care to patients with SMART This guide presents articles and materials on the legal basis for the provision of medical care and drug provision to patients, requirements for the preparation of medical documentation and its forms, and answers to the most frequently asked questions. SMA Families Foundation
Intellect in SMA disease remains completely intact; it develops in the same way as in healthy people. Thus, despite physical limitations, children can live a full life: communicate, play, do physical therapy, and go for walks.
At the moment the disease is incurable. Specialists from different countries are working on drugs to treat SMA, but so far there are no publicly available drugs that can completely cure the disease.
At the end of 2021, the first drug for the treatment of SMA, Spinraza (Nusinersen), appeared in the world. This medicine can slow the progression of the disease, and in some cases improve the condition, but still does not cure the disease completely. At the moment, Spinraza has not yet been registered in Russia - this is a matter for the near future.
In addition, several innovative drugs for the treatment of SMA are currently in development and are at the stage of clinical trials. Detailed and up-to-date information about this can be found on the website of the SMA Families Foundation in the “Research” section.
Nevertheless, we can already do a lot, and with the help of symptomatic therapy and a variety of supportive techniques, slow down the progression of the disease and, in some cases, prevent the development of complications.
Standard Treatments
There is no medicine that can cure Werdnig-Hoffmann disease. Treatment is aimed at specific symptoms that are present in each patient. Treatment may require a team of specialists.
Nusinersen is a prosurvival motor neuron 2 (SMN2) antisense oligonucleotide approved by the FDA for the treatment of SMA in adults and children. It is administered intrathecally. It increases the inclusion of exon 7 in SMN2 messenger ribonucleic acid (mRNA) and promotes the production of full-length SMN protein.
- Supportive treatment.
Medicines that are often used to improve symptoms include phenylbutyrate, valproic acid, albuterol, and hydroxyurea. Unfortunately, clinical trials have not shown any definitive evidence that these medications prevent progression of the disease. Treatment is aimed at controlling symptoms. Symptomatic treatment focuses on support with feeding, breathing and motor weakness.
Feeding problems.
Children often have difficulty feeding and may have malnutrition or aspiration pneumonia caused by difficulty swallowing. Percutaneous endoscopic gastrostomy (PEG) tubes can help with nutrition.
Breathing problems.
Children may require non-invasive ventilator support initially as the disease affects the respiratory muscles. As their symptoms worsen, they may require a tracheostomy and mechanical ventilation.
Motor weakness.
Physical and occupational therapy can help stretch, strengthen muscles, and minimize contractures. Surgical procedures and braces to help with scoliosis may be helpful.
Diet for spinal amyotrophy
At this time, no diet has been proven to be beneficial for SMA.
According to a large number of parents, a diet that includes a lot of protein or special food additives can increase the strength of the child's muscles. But, despite the obvious need for good nutrition for a sick child, it has not yet been proven that he needs a specific diet. Moreover, some products can even harm his body.
For example, the amino acid menu is sometimes fraught with even greater problems for those children who have too little muscle tissue in their bodies. According to some experts, if there is a lack of muscle tissue, it cannot properly process amino acids and then their level in the blood rises too much.
Doctors have not proven that any diet will improve the condition of a patient with SMA, but proper nutrition can make his life easier.
Some children find it healthier to eat small amounts, more often than three or four times a day. You just need to divide for the patient the entire amount of food taken by a healthy peer of the patient per day into several parts.
Principles of treatment of spinal amyotrophy
Unfortunately, this is an incurable hereditary disease. At the present stage, research is being conducted that may help regulate the synthesis of the SMN protein, but there are no results yet.
The following help alleviate the condition of patients with spinal amyotrophy:
- periodic course intake of drugs that improve the metabolism of nervous tissue and muscles (Cerebrolysin, Cytoflavin, Glutamic acid, ATP, Carnitine chloride, Methionine, Potassium orotate, Tocopherol acetate, etc.);
- B vitamins (Milgamma, Neurovitan, Combilipen);
- anabolic steroids (Retabolil, Nerobol);
- agents that improve neuromuscular conduction (Proserin, Neuromidin, Galantamine, Dibazol);
- massage and physical therapy courses;
- physiotherapy (electrical muscle stimulation, carbon sulfide baths);
- methods of orthopedic correction (with the development of joint contractures and spinal deformities).
Werdnig-Hoffmann spinal amyotrophy, like other forms of this disease, is a pathology that is inherited. The appearance of the disease in a child is explained by the presence of a mutant gene in both the mother and father. The disease is characterized mainly by muscle weakness, which causes immobility and respiratory problems. The disease is currently incurable.
Treatment methods
Treatment of spinal amyotrophy is symptomatic and aimed at stabilizing the patient's condition.
Medicines prescribed :
- improving metabolism - cerebrolysin, lipocerebin, aminalon;
- affecting the trophism of muscle tissue - potassium orotate, glutamic acid, methionine, tocopherol acetate;
- promoting neuromuscular conduction - prozerin, galantamine, dibazol;
- stimulating blood circulation in capillaries - complamin, nicotinic acid;
- supporting the viability of motor neurons - valproic acid, riluzole, L-carnitine.
Patients are prescribed orthopedic procedures in combination with warm baths, therapeutic exercises, gentle massage, oxygen therapy, and sulfide baths are indicated.
Diagnostics
Manifestations of proximal spinal amyotrophy often resemble the course of other neurological and congenital diseases, as well as traumatic injuries to the structures of the spinal cord and brain. Diagnosis of this disease is especially difficult in newborns and young children.
The key points in diagnosing spinal amyotrophy are the following studies:
- Careful history taking. The presence of cases of spinal amyotrophy in relatives allows us to suspect this hereditary disease.
- Electroneuromyography is a special study of the neuromuscular system. In this case, primary muscle damage is excluded and signs indicating pathology of the motor neurons of the spinal cord are identified.
- Computed and magnetic resonance imaging. These methods sometimes make it possible to detect atrophic changes in the anterior horns of the spinal cord. However, more often they are used to exclude other pathologies from the structures of the spinal column and brain.
- Muscle biopsy followed by histological examination of the biopsy sample. Specific muscle changes are revealed, consisting in the alternation of bundled atrophic and unchanged muscle fibers. In addition, compensatory hypertrophied muscle areas can be detected, as well as replacement of muscle tissue with connective tissue.
- Genetic analysis. Allows you to identify the exact cause of the disease: DNA testing reveals a gene mutation on the fifth chromosome.
If there have been cases of the birth of children with spinal amyotrophy in the family, when planning a subsequent pregnancy, the married couple is sent for consultation with a geneticist. Prenatal fetal DNA testing is also mandatory. Detection of Werdnig-Hoffman syndrome at the stage of prenatal diagnosis serves as an indication for termination of pregnancy.
Diagnosis and treatment of Werdnig-Hofmann disease
In the early stages of the disease, it can be difficult to differentiate the disease, since the symptoms may be similar to other diseases:
- acute poliomyelitis is characterized by the absence of disease progression and asymmetrical paralysis;
- myopathy – also of hereditary origin, has a progressive course, but the cause of muscle weakness is a violation of metabolic processes in them;
- congenital myatonia is most similar to Werdnig-Hoffmann disease; they can be distinguished quite easily using a biopsy of muscle tissue.
To diagnose the disease, a neurologist will need data on the first manifestation of symptoms, the nature of their development, and the presence of concomitant diseases.
A number of studies are carried out to make a diagnosis:
- Electroneuromyography reveals disturbances in the functioning of the neuromuscular system. Changes in the muscle type are observed, which indicates pathology of the motor neutron;
- Genetic analysis reveals a mutation in the SMN gene;
- Blood biochemistry for the level of creatine kinase, indicators within the normal range do not exclude the disease;
- Muscle biopsy for morphological examination, which reveals fascicular atrophy of muscle fibers alternating with healthy ones, as well as proliferation of connective tissue;
- MRI to rule out other diseases.
To diagnose the fetus in utero, chorionic villus biopsy, cordocentesis, and amniocentesis are used. Detection of the disease is an indication for termination of pregnancy. It is impossible to cure a patient with Werdnig-Hoffman disease. To prolong life and improve its quality, symptomatic treatment is used. The development of the disease and the worsening of symptoms are restrained by ensuring the functioning of metabolic processes in muscle tissue.
With the help of physical therapy and massage, blood circulation is improved, the risk of congestion is reduced, muscle performance is maintained, and joint immobility and loss of elasticity are prevented. Loads should be short and careful. Physiotherapy helps to maintain motor skills at the existing level and strengthen them. Special devices will help you move independently, use a computer, and even write. Portable ventilators enable patients to stay outside the hospital and live their lives more productively.
Spinal amyotrophy of Werdnig-Hoffmann - is this disease curable?
Spinal amyotrophy Werdnig-Hoffmann is a hereditary pathology of the nerves that affects the part of them that controls the skeletal muscles. It is characterized by weakness of all muscles in the body at once. More than half of the nerve cells that control muscles are located in the spinal cord.
That is why the disease is called “spinal”. It is also called “muscular” because of the detrimental effect on the muscles: they do not receive any signals from these same nerves. "Atrophy" is a medical term that refers to the wasting or shrinking of something that is not used. Here it concerns inactive muscles.
A person with such a disease has no ability to sit, move, or even take care of himself. There is no cure for this. By conducting prenatal diagnostics, you can prevent the birth of a baby with this disease.
Here we will talk about how this pathology is inherited, what its manifestations are, and also about how you can help a sick person.
We advise you to study - Lordosis of the cervical spine: treatment, symptoms, diagnosis and causes of the pathological disorder
Spinal amyotrophy Werdnig-Hoffmann is named after the two scientists who first described it. In the second half of the 19th century, they proved the morphological essence of the disease. At first, both scientists believed that this pathology had only one form.
But already in the 20th century, scientists Kueckelberg and Welander discovered another clinical form of it, with a genetic cause similar to that discovered by Werdnig and Hoffman. Several clinical forms of spinal amyotrophy are now known.
They are united by a common hereditary defect.
This pathology is hereditary. It is based on a mutation on chromosome 5. The gene that produces the SMN protein is mutated. This protein is necessary for motor neurons to develop exactly as needed.
If the fifth chromosome mutates, it will negatively affect motor neurons, interfering with their development, or even completely destroying them. As a result, the muscle cannot receive control signals from the nerves, and therefore cannot function.
It turns out that not a single movement associated with it is performed.
The mutated gene has an autosomal recessive mode of inheritance. The phrase is deciphered as follows: for the development of spinal amyotrophy, it is necessary that both parents have a mutant gene.
To put it simply, the disease will not develop if at least one of the parents was not a carrier of the mutated gene. At the same time, they themselves do not get sick: people have paired genes, and the healthy gene dominates in the father and mother of the child.
In this case, a sick baby is born in about a quarter of cases. Scientists estimate that about 2% of living people are carriers of a gene with such a mutation.
Classification
Three types of this pathology are known.
- The most severe, manifesting itself earlier than others.
- Medium-heavy.
- The mildest, manifesting itself at a very late age.
According to some doctors, there is another type: moderate/mild SMA, which manifests itself in an adult.
It is worth noting that in addition to Werdnig-Hoffmann spinal amyotrophy, there are other types of SMA, which differ in symptoms and types of inheritance. They are listed in the table below.
| SMAX1 | X-linked recessive | It is observed mainly in the elderly, affects the bulbar nerves of the skull, causing descending paralysis. |
| SMAХ2 | X - clutch. recessive | Congenital aggressive form, leading to death before 3 months. Causes weakness, areflexia, contractures and fractures. |
| SMAX3 | X - clutch. recessive | It mainly affects boys. Atrophy of all distal muscles. Slow increase in symptoms. |
| Distal DCMA1 | Autosomal - recessive | Congenital, mainly affects the hands, and severe respiratory disorders are possible. |
| Distal forms DCMA2 - DCMA5 | Autosomal - recessive | All four forms are characterized by slow progression; DCMA5 is diagnosed in young people. |
| Juvenile SMA (HMN1 type) | Autosomal dominant | Occurs in youth |
| Congenital spinal amyotrophy | Autosomal dominant |
Causes
Spinal muscular atrophy is associated with a genetic mutation in the SMN1 gene.
Human chromosome 5 contains two nearly identical genes at location 5q13: a telomeric copy of SMN1 and a centromeric copy of SMN2. In healthy people, the SMN1 gene encodes a motor neuron protein that, as its name suggests, plays a critical role in the survival of these cells.
In people affected by SMA, the SMN1 gene is mutated in such a way that it cannot properly encode the SMN protein - this is due to the "excision" of some coding fragments, or due to other point mutations (often resulting in a functional conversion of the SMN1 sequence to SMN2). However, almost all people have at least one functional copy of the SMN2 gene (most have 2–4 of them), which still encodes small amounts of SMN protein—about 10–20% of normal levels. This is why some neurons still survive.
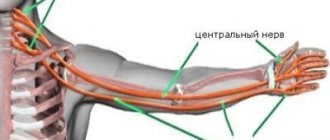
However, in the long term, reduced availability of SMN protein leads to the gradual death of motor neuron cells in the anterior horn of the spinal cord and in the brain. Muscles that rely on these motor neurons have reduced innervation (also called denervation) and therefore have much less control from the central nervous system.
A decrease in impulse transmission through motor neurons leads to a decrease in the contractile activity of the denervated muscle. Consequently, denervated muscles undergo progressive atrophy due to lack of engagement in movement.
The muscles of the lower extremities are usually the first to be affected, followed by the muscles of the upper extremities, spine and neck, and in more severe cases the pulmonary and masticatory muscles.
The severity of disease symptoms is generally related to how well the remaining SMN2 genes can compensate for the loss of SMN1 function. This is partly due to the number of copies of the SMN2 gene present on the chromosome. While healthy people carry two copies of the SMN2 gene, people with SMA can have between 1 and 4 copies of the gene, with the higher the number of SMN2 copies, the less severe the disease. Thus, most children with SMA type I have one or two copies of SMN2; people with SMA II and III usually have at least three copies of SMN2; and people with SMA IV usually have at least four copies. However, the correlation between symptom severity and SMN2 copy number is not absolute, and there appear to be other factors that influence disease severity.
Spinal muscular atrophy is inherited in an autosomal recessive manner. This means that the defective gene is on an autosome (non-sex chromosome). Two copies of the defective gene (one from each parent) are required to inherit the disorder: parents can be carriers and not show signs of the disease. Due to mutations without obvious inheritance from parents, the disease appears in only 2-4% of cases.
Affected siblings usually have a very similar form of the disease. However, different types of SMA occur among siblings, which is very rare.
What is SMA?
SMA (spinal muscular atrophy) is a genetic neuromuscular disease that affects the motor neurons of the spinal cord and leads to increasing muscle weakness. The disease is progressive, weakness begins in the muscles of the legs and the whole body and, with the development of the disease, reaches the muscles responsible for swallowing and breathing. At the same time, the intelligence of patients with SMA is absolutely preserved.
Depending on the severity of symptoms, there are 3 main types of proximal SMA: SMA 1, SMA 2, SMA 3. The earlier the first signs of the disease appear, the more pronounced the symptoms, the more severe they are and the faster the disease progresses.
SMA I (WERDNIG-HOFFMAN DISEASE)
The most severe form. Age of manifestation of the disease: up to 6 months.
Description
- Severe muscle hypotonia; "floppy child" syndrome; can't hold his head up; does not achieve the ability to sit and roll over; saggy body when held suspended on the stomach;
- Weakened cough, sucking and swallowing reflexes; choking; respiratory disorders;
- There may be a history of decreased intrauterine activity of the fetus. Deformation of joints and limbs may occur due to intrauterine hypotension.
Flow
- Severe delay in motor development;
- Rapid development of contractures and deformities of the chest;
- Progression of bulbar and respiratory disorders, problems with swallowing food and saliva, sputum discharge;
- High risk of developing aspiration pneumonia;
- Rapid increase in respiratory failure, especially when an infection occurs.
Forecast
- The most severe form: in the absence of respiratory support, most children do not survive beyond 2 years of age;
- Death occurs, as a rule, due to increasing respiratory failure and the development of pneumonia;
- Timely respiratory support can increase a child's life expectancy;
- Such children require palliative care.
SMA II (DUBOVITZ DISEASE)
Age of manifestation of the disease: 6-18 months.
Description
- Delayed motor development;
- The ability to sit without support, sometimes to crawl or stand, but these abilities are reduced as they grow older;
- Finger tremor may occur;
- Muscular and skeletal deformities;
- Breathing disorders.
Flow
- Delayed motor development, its stop and regression;
- Weakness of the intercostal muscles, shallow diaphragmatic breathing, weakening of cough function, and over time the development of respiratory failure;
- Increased risk of complications after a respiratory infection;
- Chest deformities, contractures, scoliosis.
Forecast
Timely care and respiratory support increase life expectancy.
Yulia Samoilova. The most famous person with SMA in Russia. Photo: https://www.instagram.com/jsvok/
SMA III (KUGELBERG-WELANDER DISEASE)
Age of onset of disease: after 18 months
Description
- Ability to walk independently (loses over time);
- Difficulty with complex motor skills (eg, climbing stairs, running);
- As the disease progresses, you may experience difficulty chewing and swallowing, as well as breathing and coughing problems.
Flow
- Progresses slowly;
- By adolescence, most patients use a wheelchair, but some may retain the ability to walk independently into adulthood;
- Over time, pronounced contractures and scoliosis appear;
- Risk of complications after respiratory infection.
Forecast
With proper care they have a normal lifespan.
Symptoms
To date, 4 forms of spinal amyotrophy are known. They all differ in the period of onset of the disease, some symptoms and life expectancy. Common to all forms is the absence of sensory and mental impairment. The functions of the pelvic organs are never affected. All symptoms are associated only with damage to the motor sphere.
Spinal amyotrophy type I
The onset of the disease before the age of 6 months has an extremely unfavorable prognosis.
There may be disturbances in sucking and swallowing, and difficulty moving the tongue. Fasciculations may be visible on the tongue itself (involuntary muscle contractions, “waves” running across the tongue), and it itself looks atrophied. The baby's cry is sluggish and weak. If the pharyngeal reflex decreases, problems with feeding arise, resulting in food entering the respiratory tract. And this causes aspiration pneumonia, from which the child can die.
Damage to the diaphragm and intercostal muscles is manifested by impaired breathing. Initially, this process is compensated, but gradually respiratory failure worsens.
It is characteristic that the facial muscles and muscles responsible for eye movements are not affected.
Such children are lagging behind in motor development: they do not hold their heads up, do not roll over, do not reach for objects, and do not sit up. If some motor skills were able to be realized before the onset of the disease, they will be lost.
In addition to movement disorders, the disease is characterized by chest deformation.
If signs of the disease are visible immediately after birth, then such children often die within the first 6 months of life. If signs appear after 3 months, then the lifespan is slightly longer - about 2-3 years. Inevitably, an infection occurs due to respiratory problems, from which such children die.
Spinal amyotrophy can be combined with congenital malformations: mental retardation, small skull, heart defects, congenital fractures, hemangiomas, clubfoot, undescended testicles.
Spinal amyotrophy type II
This form of the disease occurs between the first 6 months and 2 years of life. Before this, no violations are detected in the child. He begins to hold his head up, roll over and sit, and sometimes walk. And then muscle weakness gradually appears. It usually starts with the thigh muscles. Walking gradually becomes impossible, tendon reflexes decrease and are lost. Muscle weakness progresses slowly. All limbs are involved. Muscle atrophy develops. The process can also involve the respiratory muscles. Also, as with type I spinal amyotrophy, facial muscles and eye muscles are not affected. There may be trembling of the hands, twitching of the tongue and limbs. Weakness of the neck muscles is manifested by drooping of the head.
Osteoarticular deformities are very characteristic: scoliosis, funnel chest, dislocation of the hip joint.
This form has a more benign course than spinal amyotrophy type I, but most patients have respiratory problems by adolescence. Poor chest excursion contributes to the development of infections, which can kill the child.
Spinal amyotrophy type III
This form is described by Kuckelberg and Welander. It is considered juvenile spinal amyotrophy. The onset of the disease is between 2 and 15 years.
The first symptom is always unsteady walking due to increasing weakness in the legs. The tone in the legs decreases, muscle atrophy develops (the muscles become thinner), but this is not always noticeable due to the well-developed layer of subcutaneous fat at this age. Children stumble, fall, and move awkwardly. Gradually, movements in the legs become impossible, and the patient stops walking.
Gradually, the disease also affects the upper limbs; the hands are affected later. With this form, weakness of the facial muscles develops, but eye movements are preserved in full. There are no reflexes from those muscle groups that are already involved in the process.
Skeletal deformities are also characteristic: funnel chest, joint contractures.
This form of the disease, with maintenance therapy, allows patients to live up to 40 years.
Spinal amyotrophy type IV
This form of the disease is considered “adult” because it appears after 35 years. Weakness also occurs in the leg muscles, decreased reflexes, and muscle atrophy, which ultimately leads to a complete loss of movement in the legs. In this case, the respiratory muscles are not involved in the process, and there are no breathing problems. Life expectancy in this form of the disease is almost the same as in healthy people. The course is the most benign compared to other forms.
Diagnostics
In the early development of the disease, it can be difficult to make an accurate diagnosis, since the symptoms are similar to other diseases. First of all, the child should undergo a consultation with a neurologist. If the baby has a disease at birth, then an approximate diagnosis can be made in the maternity hospital. The specialist conducts an examination and checks for violations of the motor system.
The following studies are being carried out:
Magnetic resonance imaging is ordered to check the spine. It is necessarily used for spinal amyotrophy, because it allows you to understand the state of the area of interest. The procedure is considered safe because it does not worsen a person’s health. Moreover, it is recommended even for children who have Werdnig's amyotrophy. If specialists and parents understand that the minor will not be able to lie still for about an hour, then the question of using anesthesia may arise. In any case, you should not refuse an MRI; it will allow you to learn a lot of useful information about your health status and the development of Werdnig’s amyotrophy.
Electroneuromyogaffy helps to study the state of nerve and muscle endings. This is also required in order to understand how severe the disease is.
If a person has spinal amyotrophy, then it is important to collect as much information as possible about the state of the body. In particular, you will have to undergo this examination.
Genetic diagnostics makes it possible to identify gene mutations. This is relevant for cases where Werdnig's amyotrophy is present. Of course, the examination is not the simplest, and not all clinics can carry it out. Moreover, it is mandatory for those people who are faced with spinal amyotrophy.
Congenital pathology can be detected before the baby is born. Diagnosis is carried out if a girl experiences weak fetal movement. Then the pregnant woman should go to the hospital for a full examination.
Fazio-Londe disease
This is a special variant of the manifestation of atrophy. Pathology begins to develop, as a rule, by the age of three, and in some cases in adolescence. The disease is characterized by weakness of the facial muscles, including the masticatory muscles. There is difficulty swallowing and voice changes. The pathology is accompanied by atrophy of the tongue, and in some cases ophthalmoplegia may appear. The disease progresses very quickly. After 6-12 months, death occurs. Paralysis and paresis in the limbs may be added to bulbar disorders. In some cases, these symptoms do not even have time to develop. However, an autopsy always reveals a lesion in the cells of the anterior spinal horns along its entire length.
Causes and factors of the disease
Why does spinal amyotrophy appear? What risk factors can be identified? What is the main cause of the disease? The disease is genetic in nature and is transmitted in an autosomal recessive manner. In this case, both parents must have the defective gene. In this case, spinal atrophy occurs in the child in 25% of cases.
How do genes influence the development of the disease? Spinal amyotrophy occurs when there is a deficiency or complete absence of the SMN protein. It ensures the survival of motor neurons. Its deficiency is the main cause of muscle atrophy in SMA. Brain cells die and there is no signal from them to the muscles.
When the long arm of chromosome 5, on which the SMN 1 gene is located, is deleted, the protein is not produced. Spinal amyotrophy develops.
Risk factors:
- Family history is significant for stillbirths;
- The disease was detected in close relatives;
- Familial infant mortality cases;
- Spinal amyotrophy in an older child.
The likelihood of muscle atrophy occurring in younger children is 1:4.
What do you need to remember?
- Spinal amyotrophy is a rare genetic pathology that leads to disability and death of the patient.
- The main cause of muscle atrophy is a genetic mutation.
- Type I disease - Werdnig-Hoffmann amyotrophy is the most severe and frequent amyotrophy, leading to the death of a child.
- The main diagnostic method is genetic testing to detect the defective gene.
- This disease is incurable.
- The prognosis for recovery and quality of life is unfavorable.
Literature
- A.N. Baklanov, S.V. Kolesov, I.A. Shavyrin. Surgical treatment of severe neuromuscular scoliosis in patients suffering from spinal muscular atrophy // Spine Surgery. - 2011. - No. 3.
- Seliverstov Yu.A. Spinal muscular atrophy: concept, differential diagnosis, treatment prospects / Yu.A. Seliverstov, S.A. Klyushnikov, S.N. Illarioshkin // Nervous diseases. - 2015. - No. 3.
- Hardart MK Spinal muscular atrophy-type I /MK Har-dart, RD Truog//Arch. Dis. Child. - 2003. - Vol. 88, no. 10.
- Wilton NC Spinal muscular atrophy: the challenges of “doing the right thing” / NC Wilton // Paediatr. Anaesth. - 2009. - Vol. 19, no. 11.
- The changing natural history of spinal muscular atrophy type 1 / M. Oskoui, G. Levy, CJ Garland // Neurology. - 2007. - Vol. 69, no. 20.
- Anesthesia and perioperative medical management of children with spinal muscular atrophy / R..J. Graham, U. Athiraman, A. E. Laubach // Paediatr. Anaesth. - 2009. - Vol. 19, no. 11.
Types of spinal amyotrophies
Spinal muscular atrophy is one of the most dangerous genetically determined diseases that is found in infants, adolescents, and adults.
It's scary to find out that the baby will never sit, stand, or run. It’s even more scary to see how a normally growing and developing child suddenly begins to slowly fade away, constantly fall, after a few months cannot climb the stairs, and one day loses the ability to simply stand up.
Conventionally, proximal and distal forms of SMA are distinguished. 80% of all types of spinal amyotrophy are of the proximal form.
These include, in addition to Werdnig-Hoffmann disease:
- SMA 3 or Kuldberg-Welander disease - occurs between the ages of 2 and 20, and the pelvic muscles are the first to suffer. There is tremor of the hands and lordosis.
- Lethal X-linked form - described in 1994 by Baumbach, is inherited in a recessive manner, predominantly affecting the muscles of the pelvis and shoulder girdle.
- Infantile degeneration - reflexes of sucking, swallowing, breathing are impaired. Death may occur before the age of 5 months.
- SPA Ryukyu - the linkage gene has not been identified, there is a lack of reflexes, muscle weakness of the limbs after birth.
Distal spinal amyotrophies include progressive Fazio-Londe paralysis, Brown-Vialetta-van Laere disease, SMA with diaphragmatic paralysis, epilepsy and oculomotor disorders.
Prognosis: How long do patients live?
Spinal amyotrophy is an extremely serious disease that significantly worsens the quality of life of patients and is one of the causes of infant death. Patients with this diagnosis are disabled and are often unable to care for themselves.
Many patients cannot walk or stand using a wheelchair. When the hands are affected, patients are unable to do basic things: eat, hold small objects, read, wash, etc. They need additional care and constant medical supervision. In severe cases, they breathe only with the help of a ventilator and are fed through a tube.
Life expectancy largely depends on the form of the disease. The most severe is Werdnig's amyotrophy, in which affected children rarely reach 4 years of age. Patients with the second type of disease rarely survive to adulthood. The most favorable are the second and third types of the disease, in which patients reach adulthood.
Currently, spinal amyotrophy is an incurable disease and often leads to death.
Clinical picture
The symptoms of SMA 1 and SMA 2 are very different. Manifestations of SMA 1 are often detected during pregnancy, since the fetus in the womb is inactive and moves very rarely. After birth, the child experiences respiratory failure, often he cannot breathe on his own, and other innate reflexes are absent. The main manifestations of the disease can be considered:
- Decreased muscle tone.
- Retarded physical development.
- Inability to hold the head upright.
- Cannot independently roll over onto its side, stomach or back.
- Mild paralysis of the limbs.
- Lack of swallowing reflexes.
- Lack of sucking reflex.
The child takes a characteristic position in which he is almost constantly. In most cases, paralysis of the diaphragm may also occur. All this over time leads to disruption of skeletal development, scoliosis appears, the presence of a hump is often noted, and the shape of the chest changes. Only 12% of such children live to be 5 years old.
Type I amyotrophy
The most malignant and common type of pathology is Werdnig-Hoffmann amyotrophy. Diagnosed in young children or in utero. After birth, children experience a decrease and extinction of all reflexes. They find it difficult to breastfeed, which often makes it necessary to feed the baby through a tube.
Muscular hypotonia leads to weakness of the neck muscles - children cannot learn to hold their heads, roll over independently, sit, stand and walk. Most children have difficulty swallowing. In most cases, amyotrophy is diagnosed within 6 months after birth. Parents pay attention to the child’s lethargy and inactivity, and a progressive decrease in muscle tone. Babies also have trouble gaining weight.
It is characterized by damage to the diaphragm, which is involved in the respiratory act. Many patients cannot breathe on their own. Due to severe respiratory failure, many children do not live to see one year of age. It is possible to prolong their life with the help of mechanical ventilation and feeding through a feeding tube. 95% of children die before age 4.
The pathology is accompanied by progressive deformations of the musculoskeletal system.
In addition to damage to muscles and bones, mental retardation is often detected.
Reference. The diagnosis can be suspected even during the mother's pregnancy, when there is weak fetal movement, cardiac dysfunction, and developmental delays.