Features of the course of Landouzy-Dejerine muscular dystrophy
The neuromuscular disease is characterized by progressive damage to the facial muscles, shoulders and shoulder blades. Muscles in other parts of the body are also affected, but much more slowly and less severely. A feature of the flow is the asymmetry of the destructive process. Often the muscles of the shoulder girdle are the first to atrophy, gradually spreading to the facial muscles, leading to facial expression, weakness and the inability to raise the arms.
Unlike other muscular dystrophies, with Landouzy-Dejerine myopathy, sports and physical activity do not slow down the pathological process, but rather accelerate the destruction of muscle tissue. In the future, cardiac dysfunction is observed, vision loss and hearing loss may occur.
This myopathy was first described by the French doctors Landouzy and Dejerine in 1884.
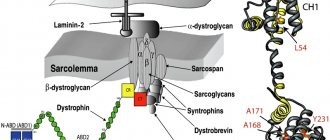
It is known that the diagnosis is genetically determined, with an autosomal dominant mode of inheritance. Most patients have gene mutations, but some patients do not have genetic disorders.
Landouzy-Dejerine glenohumeral-facial myopathy is a rare pathology, with an incidence rate of 1-1.5 per 100,000 population. Due to hereditary predisposition, it is often detected in blood relatives. The first symptoms appear in childhood. The peak incidence and severity of clinical manifestations are adolescence, pregnancy and menopause.
The pathology continues to be studied, but the cause of the defect and the development of atrophy is not known. There is currently no pathogenetic treatment, which is why the disease continues to be classified as a serious disease. Despite the fact that patients remain able to work for a long time and can care for themselves, in most cases the diagnosis leads to their disability.
Organizations and communities dedicated to FSHD. Registers of patients with FSHD
On the TREAT-NMD website you can find detailed information about international organizations involved in Landouzy-Dejerine Facioscapulohumeral Muscular Dystrophy: https://www.treat-nmd.eu/FSHD/overview/. You can also find international patient registries .
Why do you need to register in the registry?
As new drugs are developed, there is a need to test them in clinical settings, and sometimes it takes years to find the right number of patients for studies, since muscular dystrophies are a rare (orphan) disease.
For this purpose, patient registries (registers) are maintained in different countries - databases of genetic and clinical information about people suffering from neuromuscular diseases and who want to speed up the research process. The register allows specialists to obtain information about the condition and number of patients with a certain disease. This information contributes to the development and improvement of patient care standards. It is used to find participants for clinical trials and also to help specialists gain more information about a disease.
Reasons for development
The primary causes of the biochemical defects leading to myopathy are unknown. But, analyzing statistical data, scientists identify several factors in the development of myopathy:
- hereditary factor - often members of the same family are affected;
- genetic predisposition - many have a defect in chromosome 4;
- viral diseases in the mother during pregnancy;
- pathology of pregnancy in the mother.
In some patients, myopathy occurs against the background of normal health for no apparent reason. In this case, the diagnosis is classified as idiopathic myopathy.
Due to the lack of information about the initial processes of pathology development, doctors are practically unable to stop them and restore lost body functions.
Symptoms
The first signs of the disease appear at 10-20 years of age, when excessive muscle weakness, fatigue and slow development of the muscles of the shoulder girdle are noted. Patients find it difficult to raise their arms and perform physical exercises. Due to atrophy of muscle tissue, their shoulder blades protrude excessively, acquiring a wing-shaped shape, the gap between them is widened, and the chest is flattened. Muscular weakness contributes to the development of scoliosis.
Dystrophy of the muscles of the shoulder, chest and trapezius muscles leads to the development of the symptom of a drooping shoulder. A common complication in this case is dislocation of the shoulder joint. Patients experience difficulties in everyday life and at home. It is difficult for them to comb their hair, wash their face, and brush their teeth. Due to previously habitual loads, they get tired too quickly, to the point of being unable to perform them.
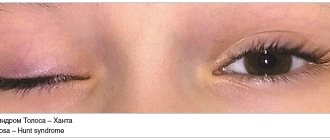
As myopathy progresses, it involves the facial muscles in the process. The face becomes amicable. Due to damage to the orbital muscles of the mouth and eyes, it is difficult or impossible to close your eyes and purse your lips. This leads to frequent eye inflammation and corneal injury.
A common complaint is speech impairment - unintelligible, slow. This significantly impairs the quality of life, especially social life and work activity. Difficulties also arise during meals. Patients have difficulty eating solid food, chewing it and swallowing it.
Characteristic signs of the disease are: inverted lips “tapir lips”, a transverse smile “Gioconda’s face”, a polished forehead.
In the future, progressive muscular dystrophy in other parts of the body is possible. When the tissues of the buttocks and thighs are affected, muscle weakness contributes to excessive fatigue from walking, which can lead to lameness. Damage to the calf muscles is accompanied by the symptom of foot drop, severe weakness when walking and the inability to run.
Depending on which symptoms are more pronounced and what is the order of their occurrence, the following forms of Landouzy-Dejerine myopathy are distinguished:
- facioscapulohumeral;
- facioscapular-humeral-peroneal;
- facioscapulohumeral-gluteofemoral;
- facioscapulohumeral-gluteal-femoral-peroneal;
- facioscapulohumeral-peroneal-gluteofemoral;
- infantile form.
The most severe is the infantile form. It occurs in children under 5 years of age. It is characterized by symmetrical paresis of the facial muscles, damage to the shoulders, chest, and absence of tendon reflexes. Due to rapidly increasing weakness, children experience respiratory failure up to the need for artificial ventilation.
Important! Often, parents of children with excessive muscle weakness try to restore the body with the help of sports sections and physical activity. As a result, the disease progresses even faster.
[edit] Clinical picture
In the classic version, the first signs of the disease begin to appear in adolescence (about 10-17 years). Usually the initial manifestations are symptoms such as difficulty raising your arms above your head, combing, and it becomes difficult to endure habitual physical activity. Visually, the following become pronounced: protruding “wing-shaped” shoulder blades and scoliosis.
Atrophy and muscle weakness are localized in the area of facial muscles, shoulder blades and shoulders. First, atrophy is observed in the shoulder girdle, with subsequent spread to the face.[1]
As the process progresses, the orbicularis muscles of the mouth and eyes suffer severely - it is not possible to close the eyes tightly and purse the lips. Due to atrophy, the face becomes hypomimic. Very often, patients themselves note a change in their facial expressions; their speech gradually becomes unintelligible. Pathognomonic symptoms include such symptoms as a transverse smile (“Gioconda smile”), everted lips (“tapir lips”) and a “polished” forehead. Atrophy of the biceps and triceps muscles of the shoulder, pectoralis major, serratus anterior, and trapezius muscles can cause symptoms of loose shoulder girdles, “wing-shaped” shoulder blades, the appearance of a wide interscapular space, flattening of the chest and scoliosis. In some cases, atrophy can spread to the leg muscles. In this case, weakness is most noticeable in the peroneal muscle group along the foot drop, but can also be present in the proximal parts of the legs.
In the early stages of the disease, muscle tone is reduced in the proximal muscle groups, deep reflexes are reduced mainly in the biceps and triceps brachii muscles.
It is necessary to note the asymmetry of atrophy, which is a characteristic clinical feature of this pathology. In some cases, muscle pseudohypertrophy occurs. Contractures and retractions are moderately expressed. Cardiomyopathy occurs in rare cases. Angioretinography may reveal abnormalities of the retinal vessels.
In many cases, severe ocular manifestations include telangiectasia, edema, and retinal detachment. Coagulation of telangiectasia prevents the development of blindness. Hearing loss may also occur. The above symptoms are considered as part of the phenotypic manifestations of this pathology.
The course of the disease is in most cases relatively favorable
Diagnostic methods
Despite the frequent detection of genetic mutations during genetic testing, in some patients with severe clinical symptoms they are absent. Because of this, a genetic blood test is not the main criterion for the disease. In most cases, Landouzy-Dejerine myopathy is diagnosed on the basis of characteristic clinical manifestations, primarily damage to muscles typical of the disease.
The following types of examination are also additionally carried out:
- biochemical blood test to determine the level of creatine phosphokinase;
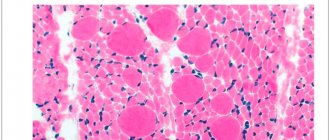
- muscle biopsy;
- electroneuromyography.
A significant increase in the level of creatine phosphokinase indicates progressive atrophy of muscle tissue. Patients have weak myocyte activity. For histological examination, tissue samples are taken from different parts of the body. The examination reveals characteristic signs of atrophy. Additionally, patients need an ophthalmological examination and consultation with a neurologist.
Important! The course of some diseases is similar to this myopathy. Doctors must carry out differential diagnosis with neurological diseases, autoimmune and connective tissue pathologies.
[edit] Diagnostics
To clarify the diagnosis, a biochemical study with the determination of creatine phosphokinase (CPK), electroneuromyography (ENMG) and a biopsy of damaged muscles are used.
The level of CPK can increase 5-7 times, but in some cases the enzyme content is normal. ENMG records both mypathic and denervation potentials.
When conducting histological studies, minimal changes are revealed in many muscles of the extremities; the largest number of pathological signs are observed in the suprascapular muscles, where phenomena of progressive degeneration are found.
Treatment
The disease is an incurable diagnosis. There is no pathogenetic treatment. It is also quite difficult to stop rapidly progressive atrophy. Treatment of Landouzy-Dejerine myopathy is aimed primarily at slowing down dystrophic changes that lead to the inability to move and care for oneself, restoring lost functions and generally improving the patient’s condition. Drug therapy is based on the use of medications that support myocytes, improve metabolic processes in them, and have a beneficial effect on the heart. The most commonly prescribed drugs are: ATP-long, Mildronate, Methion, vitamin E, Cytoflavin, cocarboxylase.
The following factors are also important for improving the condition:
- nutrition – it is recommended to increase the amount of proteins and reduce fats;
- physiotherapy – patients are prescribed massage, electrophoresis and other procedures;
- physical therapy – moderate physical activity and a special complex is necessary to prevent the development of scoliosis and contractures.
Considering that myopathy often occurs in childhood, special attention must be paid to the mental state of patients. Often the pathology leads to depressive disorders, nervous breakdowns and emotional disorders. To prevent psychological disorders and improve the emotional state of patients, they need the help of a psychologist or psychotherapist.
For socialization, it is necessary to help patients choose a profession in order to maintain their ability to work.
Watch the video on the topic of the article!
Possible complications
Despite careful study, Landouzy-Dejerine myopathy continues to be a severe pathology. In many ways, the prognosis depends on the characteristics of the course and speed of development of the pathological process. Hypomia and amymia not only limit physical capabilities, but also cause psychological discomfort and difficulties with socialization.
Progression and involvement of other parts of the body in the pathological process leads to significant restrictions, including the inability to move.
In some cases, with damage to the orbicularis oculi muscles and frequent inflammatory processes, patients' vision deteriorates to the point of blindness.
Often, following the doctor’s recommendations helps preserve life expectancy, but in extremely severe forms of the disease, death is possible due to severe respiratory failure.
What do you need to remember?
- Landouzy-Dejerine myopathy is characterized by progressive atrophy of the muscles of the face, shoulders and shoulder blades.
- The main cause of the disease is genetic predisposition.
- Signs of the pathology are hypomimicness and amymicity, muscle weakness and loss of muscle tissue.
- Treatment is aimed at inhibiting the development of the disease and preventing its progression.
- Muscular dystrophy leads to disability of the patient due to severe limitations in his capabilities.
- The prognosis for recovery is unfavorable, since the disease is incurable.
Literature
- Asanov A. Yu. Fundamentals of genetics and hereditary developmental disorders in children: Textbook. manual for university students. - M.: Academy, 2003. - P. 216.
- Badalyan L. A. Child neurology: textbook. allowance. 4th ed. - M.: MEDpress inform, 2021. - 608 p.
- Vlodavets D.V., Sukhorukov V.S., Kharlamov D.A., Belousova E.D. A method for treating congenital structural myopathies and congenital muscular dystrophies by correcting secondary mitochondrial changes: Abstract of thesis. dis. ... Ph.D. - M., 2009. - 28 p.
- Grinio L.P. Atlas of neuromuscular diseases. - M.: ANS, 2004. -167 p.
- Erokhina V. A. Rehabilitation of children with hereditary myopathies // Bulletin of the Moscow University of the Ministry of Internal Affairs of Russia. - 2015. - No. 12. - P. 304-308.
- Arvanitidis A., Henriksen K., Karsdal MA, Nedergaard A. Neo-epitope Peptides as Biomarkers of Disease Progression for Muscular Dystrophies and Other Myopathies // J Neuromuscul Dis. - 2016. - No. 30. - R. 333-346.
- Carroll MB, Newkirk MR, Sumner NS Necrotizing Autoimmune Myopathy: A Unique Subset of Idiopathic Inflammatory Myopathy // J Clin Rheumatol. - 2021. - No. 22. - R. 376-80.
- Inoue M., Nishino I. Diagnosis of Idiopathic Inflammatory Myopathy: A Muscle Pathology Perspective // Brain Nerve. - 2021. - No. 68. - R. 1431-1441.
Treatment of muscular dystrophies
Treatment is aimed at preventing or reducing the severity of damage to the joints, spine and other consequences of the underlying disease. The main task is to provide the patient with mobility for the maximum possible period of time. For this purpose, medications, physical therapy, and sometimes surgical treatment are prescribed3,4.
References
- Muscular Dystrophy Information Page. NIH (accessed 7/20/2019). URL: https://www.ninds.nih.gov/Disorders/All-Disorders/Muscular-Dystrophy-Information-Page.
- Nikitin S.S. et al. Late-onset Pompe disease: the first clinical description in Russia // Neuromuscular diseases - 2014. - No. 1.
- Zhdanova E. B., Kharlamov D. A., Belousova E. D. Somatic disorders in progressive Duchenne muscular dystrophy // Russian Bulletin of Perinatology and Pediatrics - 2011. - T. 56. - No. 5.
- Duchenne muscular dystrophy and Becker muscular dystrophy. MSD Handbook (accessed 7/20/2019).
- Belozerov Yu. M. et al. Progressive muscular dystrophy of Emery-Dreyfuss // Almanac of Clinical Medicine - 2001. - No. 4.
- Umakhanova Z. R., Zhilina S. S., Mutovin G. R. Clinical polymorphism of progressive Erb-Roth muscular dystrophy // Journal of Neurology and Psychiatry named after. CC Korsakov - 2005. - T. 105. - No. 9. - P. 48-51.
- Kirillova L. G. et al. Landouzy-Dejerine facial-brachial myodystrophy in the neuropediatrics clinic // Child's Health - 2011. - No. 1.
GZEA.PD.18.09.0435m