Общие сведения
Характерной чертой болезни является нарушение обмена веществ в скелетной мышечной ткани. Мускулы больного ребенка утрачивают функцию частично или целиком, то есть в них появляется слабость, уменьшается объем движений. Качество жизни значительно снижается. Источник: Команцев В.Н., Скрипченко Н.В., Сосина Е.С., Климкин А.В. ПОЛИНЕЙРОПАТИЯ И МИОПАТИЯ КРИТИЧЕСКИХ СОСТОЯНИЙ У ВЗРОСЛЫХ И ДЕТЕЙ: ДИАГНОСТИКА, КЛИНИЧЕСКИЕ ПРОЯВЛЕНИЯ, ПРОГНОЗ, ЛЕЧЕНИЕ // Современные проблемы науки и образования. – 2012. – № 5
Эта патология обычно имеет наследственную форму и может диагностироваться у детей любого возраста. Она не угрожает жизни, за исключением тех случаев, когда происходит атрофия сердечной мышцы и дыхательной мускулатуры. Источник: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2796972/ Chris M. Jay, Nick Levonyak, Gregory Nemunaitis, Phillip B. Maples and John Nemunaitis Hereditary Inclusion Body Myopathy (HIBM2) Gene Regul Syst Bio. 2009; 3: 181–190.
Болезнь имеет ряд осложнений:
- развитие дыхательной недостаточности;
- ограничение подвижности;
- паралич;
- застойные воспаления легких;
- депрессивный, суицидальный настрой больного;
- повышенный риск летального исхода.
Мышечная дистрофия Дюшенна: вопросы к терапии антисмысловыми олигонуклеотидами
«Амондис 45» — по аналогии с «Эксондисом 51», «Виондисом 53» и «Вилтепсо» — регуляторное одобрение получил условно: на базе суррогатной конечной точки. Четверка этих лекарственных средств обеспечила статистически значимый рост уровня белка дистрофина в скелетных мышцах, что с приемлемой вероятностью позволяет прогнозировать клиническую пользу для пациентов. FDA вынесло положительные вердикты ввиду жизнеугрожающей и инвалидизирующей природы мышечной дистрофии Дюшенна, надежной и высокоэффективной специфической терапии которой по-прежнему не предложено.
По мнению отраслевых экспертов, клинически значимой эффективностью любой терапии мышечной дистрофии Дюшенна является выход на синтез дистрофина de novo до не менее чем 10% от его нормального образования в здоровом организме. Даром что ли «Эксондис 51», обеспечивший подъем уровень дистрофина до всего лишь 0,44% после 48-недельного курса лечения, некоторые критики назвали «изящным с научной точки зрения плацебо».
К слову, Европейское агентство по лекарственным средствам (EMA) отказалось одобрять «Эксондис 51», ссылаясь на отсутствие должных клинических данных, которые бы подтвердили, что скромная прибавка уровня дистрофина, обеспечиваемая этеплирсеном, принесет пациентам долгосрочные положительные результаты. Возможно, регулятор поменяет свое решение, когда к 2026 году будут собраны результаты 144-недельного клинического исследования MIS51ON (NCT03992430) фазы III, проверяющего «Эксондис 51» в стандартной и повышенной вплоть до 6 раз дозе.
Продолжающееся клиническое испытание ESSENCE (NCT02500381), которое завершится к 2024 году, призвано подтвердить терапевтическую эффективность «Амондиса 45» и «Виондиса 53». По прошествии 96 и 144 недель лечения будет оценена мышечная функциональность пациентов: тестом 6MWT, способностью самостоятельно подниматься с пола, временем до потери амбулаторного статуса, амбулаторной оценкой состояния по шкале North Star (NSAA), показателем FVC. Кроме того, по истечении 96 недель терапии будет замерен уровень дистрофина в мышечных биоптатах (вестерн-блоттингом и иммуногистохимически).
Причины миопатии у детей:
- гормональные сбои;
- наследственность;
- генетические дефекты (дефицит фермента, обеспечивающего обменные процессы в мышцах; дефект клетки, которая играет самую важную роль в доставке энергетического материала к мышцам); Источник: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5575512/ Alessia Nasca, Chiara Scotton, Irina Zaharieva, Marcella Neri, Rita Selvatici, Olafur Thor Magnusson, Aniko Gal, David Weaver, Rachele Rossi, Annarita Armaroli, Marika Pane, Rahul Phadke, Anna Sarkozy, Francesco Muntoni, Imelda Hughes, Antonella Cecconi, György Hajnóczky, Alice Donati, Eugenio Mercuri, Massimo Zeviani Recessive mutations in MSTO1 cause mitochondrial dynamics impairment, leading to myopathy and ataxia Hum Mutat. 2021 Aug; 38(8): 970–977.
- системные поражения соединительной ткани.
Причины мышечной дистрофии
Мышечная дистрофия – это заболевание мышц, точнее, группа заболеваний, которая приводит к слабости и дегенеративным изменениям в мышцах. Дистрофия чаще затрагивает скелетные мышцы. Они постепенно слабеют, теряют способность сокращаться, уменьшаются в объеме и замещаются соединительной или жировой тканью.
Причина заболевания – мутация гена, который отвечает за синтез белков в клетках мышечной ткани. Поражению могут подвергнуться различные виды генов – как расположенные в половой хромосоме, так и не связанные с ней. От этого зависит механизм передачи заболевания по наследству (например, ген Дюшенна, связанный с половой Х-хромосомой, передается по мужской линии; женщины не болеют дистрофией, вызываемой этим геном, хотя и являются его носителями).
Симптомы и лечение патологии у ребенка
Клинические признаки миопатии у детей:
- изменение походки;
- слабость, которая не проходит после отдыха;
- задержка моторного развития;
- вялые, дряблые мышцы;
- атрофия (истончение) мускулов;
- искривление позвоночника – проявление, свидетельствующее о слабости мышечного корсета.
Негативные процессы проявляются у детей в раннем и юношеском возрасте, но так как миопатия развивается медленно, долгое время она может оставаться незамеченной. Кроме того, дети способны компенсировать мышечную недостаточность, используя более активно другие, здоровые мускулы.
Наиболее часто изменения наблюдаются в зонах плеч, ног, рук, таза, грудной клетки. Они всегда при данном недуге двухсторонни и симметричны.
С развитием болезни проявляются двигательные нарушения:
- ребенку трудно сесть из положения лежа;
- движения ненормальные, «неправильные»;
- при ходьбе и/или беге быстро наступает усталость;
- ребенок с трудом держит равновесие, часто падает;
- ребенку тяжело подниматься по лестнице.
Также могут проявиться нарушения внешнего вида:
- выступающие ребра;
- очень тонкая, будто перетянутая, талия;
- уплощенная грудная клетка;
- сутулость;
- неправильная форма ног – утолщенные икры и худые бедра.
«Амондис 45»: безопасность касимерсена в лечении мышечной дистрофии Дюшенна
Исследования на самцах мышей и крыс засвидетельствовали почечную токсичность касимерсена, проявившуюся дегенерацией почечных канальцев. И хотя испытания на людях не отметились ничем таким подобным, необходимо принимать во внимание, что в ходе изучения некоторых других антисмысловых олигонуклеотидов пациенты сталкивались с почечной токсичностью, включая потенциально летальный гломерулонефрит. Потому терапия «Амондисом 45» должна сопровождаться мониторингом функции почек.
Среди наиболее частых побочных реакций на назначение касимерсена: инфекции верхних дыхательных путей (у 65% пациентов — против 55% в группе плацебо), кашель (33% против 26%), лихорадочное состояние (33% против 23%), головная боль (32% против 19%), суставная боль (21% против 10%), боль в ротоглотке (21% против 7%).
Диагностика миопатии
Болезнь выражается:
- нарастающей симптоматикой;
- отсутствием судорог и неврологических проявлений;
- избирательной локализацией;
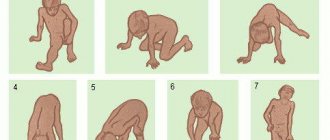
- характерной «утиной» походкой.
Для точной диагностики в первую очередь собирается анамнез, выясняется, были ли случаи этого недуга в семье. Затем проводится осмотр неврологом, в ходе которого врач оценивает мышечный тонус, распространение слабости, наличие истончения мускулов, степень деформации тела, выраженность рефлексов, походку, просит ребенка сесть из положения лежа и встать из положения сидя.
Лабораторная диагностика включает:
- клинический анализ крови;
- биопсию мышц;
- проверку уровня гормонов щитовидной железы.
Также проводится генетическое обследование ребенка и близких родственников. Источник: https://www.mda.org/disease/congenital-myopathies/diagnosis The Muscular Dystrophy Association (MDA).
Диагностика
Предельно информативным методом диагностики СМА считается генетический анализ. Его можно проводить ребенку и взрослому в любом возрасте, а с целью ранней диагностики его выполнение возможно еще на этапе внутриутробного развития. При невозможности проведения анализа ДНК и для окончательного подтверждения диагноза назначаются:
- биохимический анализ крови;
- гистологический анализ мышечных волокон;
- МРТ;
- электромиография;
- микроскопия спинного мозга;
- тандемная масс-спектрометрия.
Не стоит откладывать проведение операции на потом, так как прогрессирующие изменения способны привести к тяжелым нарушениям дыхания. Это станет абсолютным противопоказанием к хирургическому лечению. Хирурги медицинского проводят подготовительные действия, саму операцию и формируют реабилитационную систему восстановления на уровне европейских клиник по доступной стоимости.
Виды заболевания
Один из классификационных признаков – причина появления недуга. По нему выделяют миопатию:
- первичную (появляется самостоятельно при рождении, в раннем детстве или юношестве);
- вторичную (развивается на фоне других болезней).
По локализации слабости болезнь бывает:
- проксимальной (мышцы ослаблены ближе к туловищу);
- дистальной (мышцы ослаблены в конечностях дальше от туловища);
- смешанной.
Также существуют следующие формы заболевания:
- Псевдогипертрофическая (Дюшенн-Гризингера). Появляется в 3-6 лет, редко – до одного года. В основном затрагивает мышцы ног и таза. Присоединенные поражения: слабость дыхательных и сердечных мышц. Велика вероятность летального исхода еще до совершеннолетия.
- Ландузи-Дежерина. Начинается в 10-15 лет, поражает лицо. Ослабляется мимическая мускулатура, губы выпячиваются и утолщаются, нередко больной не может сомкнуть веки. Затем вовлекаются мышцы по нисходящей вплоть до плечевого пояса.
- Эрба-Ротта (юношеская). Начало болезни – 10-20 лет, в основном подвержены этой форме мальчики. Процессы проходят сверху вниз или снизу вверх, редко – сразу по всему телу или в зоне лица.
Важно!
Врожденная миопатия – одна из самых опасных форм у детей, часто заканчивающаяся летальным исходом. Ее лечение сводится к улучшению жизнеспособности и начинается уже в первые месяцы после рождения. Основное в терапии – предотвращение дыхательной недостаточности, организация зондового питания. По мере роста ребенка применяются методики ортопедической коррекции, большое значение имеет физиотерапия, социальная адаптация.
Врожденная миопатия
Болезнь центрального стержня наследуется аутосомно-доминантно, известны также отдельные спорадические случаи заболевания. Врожденная миопатия этого вида проявляется задержкой двигательного развития в период первого года жизни, реже обнаруживается у взрослых пациентов, часто сопровождается слабостью мимической мускулатуры. Характерен небольшой рост больных и хрупкая фигура, наличие скелетных деформаций. У пациентов с этим видом врожденной миопатии отмечается повышенный риск возникновения злокачественной гипертермии. В биоптате мышечной ткани выявляют мышечные волокна с единичными или множественными зонами асептического некроза.
Немалиновая врожденная миопатия включает актинопатию, небулинопатию, тропомиозинопатию и тропонинопатию. Ее наследование происходит чаще по аутосомно-доминантному принципу, но также встречается рецессивное наследование и спорадические случаи заболеваемости. Классическая форма немалиновой врожденной миопатии характеризуется синдромом вялого ребенка. Тяжелая форма проявляется еще во внутриутробном периоде в виде акинезии плода, а при рождении ребенка — тяжелыми двигательными нарушениями, слабостью мышц лица и дыхательной недостаточностью. Легкая форма этого типа врожденной миопатии диагностируется после периода раннего детства, иногда — в подростковом возрасте, и протекает без слабости лицевой мускулатуры. Существует также специфическая форма немалиновой врожденной миопатии, при которой возможно развитие офтальмоплегии, кардиомиопатии, синдрома ригидного позвоночника. Морфологическое исследование обнаруживает наличие в мышцах характерных палочко- или нитеподобных телец.
Миотубулярная врожденная миопатия чаще наследуется как аутосомная, при которой мышечная слабость выражена в легкой степени и может наблюдаться как у девочек, так и у мальчиков. Х-сцепленная миотубулярная врожденная миопатия поражает только лиц мужского пола и характеризуется более тяжелым течением со слабостью лицевых мышц, расстройством глотания и дыхательной функции. В биоптате мышечной ткани преобладает поражение волокон I типа. Отмечается центральное расположение ядер миоцитов, что соответствует мышечной ткани эмбриона на 8-10 недели беременности. В связи с эти большинство исследователей рассматривают миотубулярную миопатию как результат недоразвития мышечной ткани.
Миопатия с множественными стержнями чаще наблюдается как аутосомно-рецессивное заболевание, хотя возможен и доминантный тип наследования. Типична мышечная слабость в проксимальных отделах, которая наблюдается в грудном возрасте. Намного реже заболевание дебютирует в более старшем возрасте. В таких случаях отмечается генерализованная мышечная слабость. В мышечном биоптате определяются клетки с отсутствием митохондрий, деструкция сакромеров и гипотрофия мышечных волокон.
Врожденная диспропорция типов мышечных волокон проявляется генерализованной слабостью мышц, в том числе и лицевых, мышечной гипотонией, аномалиями скелета. Тип наследования этой врожденной миопатии пока не установлен. В биоптате мышц наблюдается увеличение количества и малый размер волокон I типа на фоне гипертрофии или нормального размера волокон II типа.
Лечебные методики
Важно!
Чем раньше начать лечить ребенка, тем больше у него шансов на достаточно высокое качество жизни.
Лечение сводится к следующим мероприятиям:
- инъекционное введение аденозинтрифосфорной кислоты (АТФ) курсами;
- ионофорез;
- витаминизация;
- препараты для улучшения кровообращения;
- массаж;
- использование больным ортопедических средств коррекции;
- применение препаратов для лучшей нервно-мышечной проводимости;
- терапия гормонами;
- и др.
Наследственная форма заболевания полностью не вылечивается, но возможно направленно устранить основные симптомы путем:
- ортопедической коррекции;
- обычной и дыхательной гимнастики.
Иногда требуется хирургическое вмешательство. Оно направлено на коррекцию сколиоза, возникающего на фоне основного недуга.
Перспективными методиками лечения миопатии являются: использование стволовых клеток и генотерапия.
Генная терапия мышечная дистрофия Дюшенна: долгая надежда
«Сарепта» продолжает разрабатывать генную терапию мышечной дистрофии Дюшенна, которая, похоже, подойдет всем больным без оглядки на мутационную подоплеку болезни. Есть надежда, что после однократной инъекции течение этого инвалидизирующего и фатального заболевания изменится кардинальным образом: пациенты не только избавятся от угрозы неминуемой смерти, но и получат шанс на более-менее полноценную самостоятельную жизнь.
Усилия «Сарепта» сосредоточены на вирус-векторной доставке в организм синтетического трансгена, кодирующего важнейшие домены белка дистрофина, критическая недостаточность которого является причиной миодистрофии Дюшенна. Поскольку аденовирусный вектор располагает ограниченной возможностью для переноса генетической информации (емкость составляет приблизительно 4,7 килобаз), он не способен транспортировать ген дистрофина, который слишком велик (2,4 мегабаз), потому вполне сойдет усеченный, но всё же функциональный вариант последнего — так называемый микродистрофин.
В начале января 2021 года «Сарепта» поделилась основными результатами продолжающегося клинического исследования NCT03769116 фазы II (рандомизированного, двойного слепого, плацебо-контролируемого, многоцентрового) генотерапевтической программы SRP-9001 (rAAVrh74.MHCK7.micro-dystrophin), охватывающего мальчиков (n=41) в возрасте 4–7 лет с мышечной дистрофией Дюшенна.
По прошествии 12 недель после однократной дозы деландистрогена моксепарвовека (delandistrogene moxeparvovec, SRP-9001) уровень экспрессии микродистрофина вышел к 28,1% (p<0,0001). Продемонстрированы также должные биологические показатели: число копий вирус-векторных геномов на ядро (1,56), пропорция (33,0%) и интенсивность окрашивания (63,7%) дистрофин-положительных мышечных волокон, снижение креатинкиназы.
По истечении 48 недель исходный общий балл NSAA в группе генной терапии увеличился на 1,7 пункта (p=0,0090) — против его роста на 0,9 пункта (p=0,1411) в группе контроля. Однако разница не оказалась статистически значимой (p=0,37).
17-компонентная оценочная шкала NSAA используется для определения функциональных моторных навыков детей с мышечной дистрофией Дюшенна и применяется для отслеживания прогрессирования заболевания и терапевтических эффектов. Шкала NSAA учитывает способности пациентов к тому, чтобы стоять, ходить, вставать со стула и пола, стоять на одной ноге, подниматься и спускаться по ступенькам, прыгать, бегать и т. п.
На фоне столь разочаровывающих известий биржевые котировки «Сарепта» потеряли сразу половину стоимости: минус 7 млрд долларов рыночной стоимости предприятия.
«Сарепта», впрочем, не расстраивается, объясняя формальный провал испытания следующим образом. Рандомизация участников была стратифицирована по возрастным группам: 4–5 и 6–7 лет. В первой группе общий балл NSAA среди прошедших генную терапию прибавил 4,3 пункта (p<0,0001) — против его роста на 1,9 пункта (p=0,0126) среди получивших плацебо. Разница в 2,5 пункта получилась статистически значимой (p=0,0172), что связано, как утверждается, с исходным функциональным статусом пациентов, который был хорошо сбалансирован между группой генной терапии и плацебо. При этом во второй группе таковой характеризовался явным расхождением: в группу плацебо фактически попали пациенты с менее тяжелой степенью выраженности миодистрофии Дюшенна, если сравнивать с больными в группе экспериментального лечения. Другими словами, терапевтический провал деландистрогена моксепарвовека среди всей популяции включенных в исследование пациентов объясняется не тем, что изучаемая генная терапия неэффективна, а ошибками с подбором испытуемых.
Как бы то ни было, клиническое испытание продолжается.
В конце декабря 2021 года «Сарепта» предоставила «Рош» (Roche) все коммерческие права на SRP-9001 за пределами США. Взамен были получены авансом 750 млн долларов наличными и 400 млн долларов акциями, обещаны дополнительные выплаты до 1,7 млрд долларов по мере прохождения регуляторных и сбытовых этапов, плюс роялти от реализации.
В генную терапию мышечной дистрофии Дюшенна также погружены «Пфайзер» (Pfizer) и «Солид байосайенсиз» (Solid Biosciences), тестирующие соответственно фордадистроген мовапарвовек (fordadistrogene movaparvovec, PF-06939926) и SGT-001.
Преимущества обращения в «СМ-Клиника»
В нашей клинике работают одни из лучших детских неврологов Санкт-Петербурга, врачи высоких категорий, обладающие внушительным опытом. Ваш ребенок сможет пройти диагностику на современном оборудовании, сдать лабораторные анализы без очередей и в комфортных условиях. Специалисты «СМ-Клиника» разработают оптимальный план лечения в короткие сроки с учетом индивидуальных особенностей пациента и формы его заболевания.
Позвоните нам, чтобы задать дополнительные вопросы и записаться на прием.
Источники:
- Команцев В.Н., Скрипченко Н.В., Сосина Е.С., Климкин А.В. Полинейропатия и миопатия критических состояний у взрослых и детей: диагностика, клинические проявления, прогноз, лечение // Современные проблемы науки и образования, 2012, № 5.
- https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2796972/ Chris M. Jay, Nick Levonyak, Gregory Nemunaitis, Phillip B. Maples and John Nemunaitis Hereditary Inclusion Body Myopathy (HIBM2) Gene Regul Syst Bio. 2009; 3: 181–190.
- https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5575512/ Alessia Nasca, Chiara Scotton, Irina Zaharieva, Marcella Neri, Rita Selvatici, Olafur Thor Magnusson, Aniko Gal, David Weaver, Rachele Rossi, Annarita Armaroli, Marika Pane, Rahul Phadke, Anna Sarkozy, Francesco Muntoni, Imelda Hughes, Antonella Cecconi, György Hajnóczky, Alice Donati, Eugenio Mercuri, Massimo Zeviani
- Recessive mutations in MSTO1 cause mitochondrial dynamics impairment, leading to myopathy and ataxia Hum Mutat. 2021 Aug; 38(8): 970–977.
- https://www.mda.org/disease/congenital-myopathies/diagnosis The Muscular Dystrophy Association (MDA).
Пицуха Светлана Анатольевна Clinic
Автор статьи
Пицуха Светлана Анатольевна
Врач высшей квалификационной категории
Специальность: невролог
Стаж: 24 года
Информация в статье предоставлена в справочных целях и не заменяет консультации квалифицированного специалиста. Не занимайтесь самолечением! При первых признаках заболевания необходимо обратиться к врачу.
Признаки
Различают несколько стадий мышечной гипотрофии:
- Во всем теле уменьшается количество подкожной клетчатки. Человек теряет до 20% массы тела. У него отмечается бледность, слабый аппетит, тонус мышц снижается;
- Подкожная клетчатка на животе и грудной клетке практически пропадает. Кожа становится серой, мышцы дряблыми, печень увеличивается в размере. Возникают расстройства психического здоровья, раздражительность;
- Резкое истощение (кахексия) возникает при потере пациентом 30% мышечной массы. Состояние требует проведения интенсивной терапии.
К общим симптомам мышечной гипотрофии относят следующие признаки:
- Постоянные боли в мышцах;
- Слабость;
- Неспособность выполнять обычные движения;
- Значительную потерю массы тела.
Если участки гипотрофии мышц располагаются симметрично, это вызывает подозрение на миопатию или спинальную амиотрофию. При прогрессирующей мышечной дистрофии наблюдается относительно изолированная гипотрофия четырехглавой мышцы бедра или двуглавой мышцы плеча. Если гипотрофии располагаются в дистальных отделах конечностей, речь идёт о полинейропатии (с нарушением чувствительности и утратой рефлексов в дистальных отделах конечностей) или миотонической дистрофии Штейнерта.
Односторонние приобретенные изолированные гипотрофии мышц всегда являются следствием поражения корешка, сплетения или периферического нерва. Решающим для топического диагноза служит характерное распределение процесса гипотрофии и нарушений чувствительности или же длительного бездействия мышцы. Гипотрофия четырехглавой мышцы возникает при артрозе коленного сустава и при саркоме бедра. Фокальные гипотрофии отдельных мышц или групп мышц, изолированные, а иногда и симметричные могут медленно прогрессировать в течение многих лет. Это является признаком очагового поражения ганглионарных клеток передних рогов или ишемии в зоне кровоснабжения артерии.
Часто возникает гипотрофия икроножных мышц. При прогрессирующей мышечной дистрофии иногда в значительно гипотрофированных мышцах определяются участки с сохранными мышечными волокнами, которые выглядят как узелки. Их врачи Юсуповской больницы отличают от мышечного валика, который образуется при разрыве короткой головки двуглавой мышцы плеча и заметен на сгибательной поверхности плеча.
Цены
| Наименование услуги (прайс неполный) | Цена |
| Прием (осмотр, консультация) врача-невролога первичный, лечебно-диагностический, амбулаторный | 1750 руб. |
| Консультация (интерпретация) с анализами из сторонних организаций | 2250 руб. |
| Назначение схемы лечения (на срок до 1 месяца) | 1800 руб. |
| Назначение схемы лечения (на срок от 1 месяца) | 2700 руб. |
| Консультация кандидата медицинских наук | 2500 руб. |
| Транскраниальное дуплексное сканирование (ТКДС) сосудов мозга | 3600 руб. |
Лечение
Неврологи Юсуповской больницы назначают пациентам, страдающим дистрофией мышц, комплексное лечение, направленное на устранение причины заболевания, оказывающее влияние на механизмы развития патологического процесса, уменьшающее проявления болезни. Для улучшения кровотока в периферических сосудах применяют ангиопротекторы (трентал, пентоксифиллин, курантил), низкомолекулярный декстран, препараты простагландина Е (вазапростан). После расширения сосудов но-шпой и папаверином улучшается снабжение мышечных волокон кислородом и питательными веществами.
Нормализуют обменные процессы и проведение нервных импульсов витамины группы В (тиамина гидрохлорид, пиридоксина гидротартрат, цианокобаламин). Стимулируют регенерацию мышечных волокон и восстанавливают объём мышц биологические стимуляторы: алоэ, актовегин, плазмол. Для восстановления мышечной проводимости применяют прозерин, галантамин, армин.
Приобретенные заболевания мышц
Приобретенные заболевания мышц могут быть воспалительного, инфекционного, лекарственного, токсического генеза.
Воспалительные заболевания мышцы человека – полимиозит, дерматополимиозит, миозит с включениями, саркоидная миопатия.
Инфекционные миозиты могут быть вирусными, бактериальными, паразитарными и др. Основные симптомы – слабость в мышцах и боли – появляются остро. Иногда могут сопровождаться поражением кожи (дерматополимиозит). Отличительной особенностью течения данных заболеваний является повышение температуры тела.