Reasons for the development of Rossolimo-Steinert-Kurshman myotonia
Rossolimo-Steinert-Kurshman myotonia is always a genetically transmitted defect. The key reason for its development is considered to be a violation in the DMPK gene, located on the nineteenth chromosome. The severity of the disease is directly proportional to the number of trinucleotide CTGs. Normally, this indicator is approximately 5-37, at 50-80 a mild form of the disease is observed, at 100-500 a late severe form is observed.
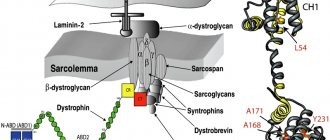
Under the influence of a violation of the DMPK gene in the body, a change occurs in myotonin protein kinase - a protein localized in skeletal and muscle tissue, in the myocardium, central nervous system, etc., which provokes the appearance of myotonic spasms, combined with atrophy of the muscles of the face, neck and limbs in the distal zones. In essence, hypertrophy of some muscle fibers occurs with simultaneous atrophy of others. As a result, some of these fibers are replaced either by other fibers or by fatty or connective tissues.
Causes of the disease
Thomsen's myotonia is based on a gene mutation in the chromosomal region that is responsible for the functioning of protein chlorine ion channels.
The mechanism of development of this phenomenon is associated with a violation of the permeability of the muscle fiber membrane, which entails the occurrence of repeated discharges in the muscle followed by a series of contractions (spasms). The instability of the cell membrane is explained by a violation of ion exchange between cells and the extracellular environment as a result of a genetic failure.
The pathology of Thomsen's myotonia is based on a gene mutation in a chromosomal region that is responsible for the functioning of protein chloride ion channels. In a healthy person, chlorine, penetrating into skeletal muscle cells, leads to relaxation of fibers and a decrease in the excitability of the cell membrane. Impaired transport of chloride ions across the cell membrane results in a series of muscle contractions (instead of just one) and prevents normal muscle relaxation as a result of electrical instability.
Symptoms and signs of classical myotonia Rossolimo-Steinert-Curschmann
The very first signs of myotonia can appear as early as the age of 6-7 years, but most often become clearly visible in adolescence and young adulthood from 10 to 20 years. The list of these signs includes myopathy, cataracts, damage to the central nervous system and cardiovascular system, and endocrine disorders.
The main symptom is the presence of muscle spasms, which mainly affect the masseter facial muscle and the flexor muscle of the hand. Also, experts always note atrophy of various muscles, including the distal parts of all extremities, temporal, sternocleidomastoid, and facial muscles. Myopathic laryngeal paresis is observed with difficulty breathing, voice changes, deterioration of ventilation, sleep pathologies and aspiration and congestive pneumonia. Due to changes in muscle tissue and premature loss of tendon reflexes, most patients experience a change in gait.
In approximately 50% of cases, patients complain of a symptom such as arrhythmia, along with which bundle branch block, as well as hypertrophy of the left cardiac ventricle, can be observed.
On the part of the SNS, the disease causes hypersomnia, a decrease in intellectual abilities, and there are often cases when patients have a mild form of debility. Due to pathologies of the endocrine system, sexual dysfunction occurs. In young women, the changes consist of the manifestation of hirsutism, menstrual irregularities and early menopause, in young people - a decrease or absence of desire and impotence, cryptorchidism and hypogonadism.
Representatives of both sexes have a common symptom of Rossolimo-Steinert-Kurshman myotonia - changes in the structure of the hair with subsequent loss. In men, baldness occurs in the area of the temples and forehead; in women, a local focal or diffuse disorder occurs.
Etiology and pathogenesis of Thomsen's myotonia
The hereditary disease is caused by a mutation in the CLCN1 gene. Etiopathogenesis includes disturbances in myoneural conductivity and pathology of intracellular membranes caused by a decrease in the permeability of the plasmalemma for chlorine ions into muscle fibers. The defect provokes an ionic imbalance: chlorine ions accumulate on the surface of the microfibril without penetrating inside, which leads to bioelectrical instability in the muscle membrane.
In the muscles of patients with Thomsen's myotonia, the level of acetylcholine is increased, and in the cerebrospinal fluid and blood there is a decrease in the activity of the enzyme involved in the regulation of the excitability and contractility of muscle fibers.
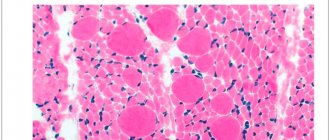
Pathomorphology does not reveal changes in muscle fibers unique to this disease. The tissue defect is typical for various types of myotonia. Light microscopy reveals hypertrophy of individual muscle fibers. Histochemistry determines a decrease in type II muscle tissue. Electron microscopy reveals moderate hypertrophy of the sarcoplasmic reticulum of muscle cells, an increase in the size of mitochondria, and expansion of the telophragm of striated muscles.
Symptoms of congenital Rossolimo-Steinert-Kurshman myotonia
The congenital type of myotonia can be noticed by a specialist even during the period of stay in the mother’s womb. This fetus does not show increased motor activity. Therefore, if a pregnant woman notices such behavior of the baby in the stomach, she should definitely undergo an ultrasound scan in the last three months before giving birth.
Signs of myotonia are also present in newborns. This is hypotonia of facial, masticatory muscles, distal areas of the limbs, and eyeballs. In addition, breathing disorders are also common. A little later, symptoms such as mental retardation and delayed motor development appear. The progression of these pathologies can occur at different rates. With the rapid pace of their development, the patient’s death from the disease can befall him at a very early age.
3. Symptoms and diagnosis
As a rule, Leiden-Thomsen disease manifests itself and can be identified diagnostically in prepubertal or early puberty. A specific symptom is residual tension (spastic tone) of any muscle groups that were tense voluntarily and adequately to the situation, after which they should relax - and do not relax. Most often, such tonic muscle spasms are observed in the fingers, hands, legs, orbicularis oculi muscles, and masticatory (masticatory) muscles. Accordingly, the patient for a long time, for tens of seconds, cannot, for example, open his eyes or mouth, turn his head, move his limbs, open his palm, change his facial expression, etc.
A characteristic feature is the often observed contradiction between athletic hypertrophy, rigidity, relief of spasmodic muscle groups and actually reduced muscle strength.
Reflexological examination using a neurological hammer easily reveals tonic-spastic reactions of the fingers, tongue, etc. Of the instrumental diagnostic methods, the most informative is electromyography. A consultation with a medical geneticist is required, and a detailed family history is collected and compared with the clinical picture.
About our clinic Chistye Prudy metro station Medintercom page!
Diagnosis of Rossolimo-Steinert-Kurshman myotonia
A specialist’s suspicion of this disease can be caused by a combination of any myotonic manifestations and dystrophic changes in muscle tissue that occur simultaneously with a lag in intellectual development, disturbances in the functioning of the endocrine and cardiovascular systems. The presence of all these signs in a patient requires confirmation of the diagnosis in the form of the results of a genealogical analysis, the results of which indicate an autosomal dominant type of genetic inheritance. In addition to these, DNA analysis data should be obtained for an accurate diagnosis. Additional studies are also needed: electromyography, electroneurography, electrocardiogram, hormonal analysis.
Determining an accurate diagnosis for suspected Rossolimo-Steinert-Kurshman myotonia requires a comprehensive study of the patient’s condition and the functioning of almost all of his organs. Therefore, specialists in genetics, cardiology, gynecology, andrology and endocrinologists can be involved in the diagnostic process.
When diagnosing, you need to pay attention not only to those changes that appear externally and are characteristic of Rossolimo-Steinert-Kurshman myotonia, but also to data on muscle biopsy and electromyographic studies.
During a biochemical analysis, the muscle enzyme and its level in tissues are studied, which is always elevated in this disease. A prerequisite for making an accurate diagnosis is the antenatal diagnosis of myotonia using amniocentesis.
Thomsen/Becker myotonia in a teenager playing sports
Myotonia (miotonia from the Greek mio small + tonia tone - small tone) is a neuromuscular disease characterized by the presence of muscle hypertrophy and the myotonic phenomenon - slow relaxation of the muscle after its contraction (the contracted muscle does not relax for a long time and then relaxation occurs extremely slowly) [ 1]. There are no pronounced morphological changes in the nervous and muscle tissues in myotonia. The disease develops slowly, and deterioration often occurs after hypothermia, mental stress, or fatigue.
This disorder was first described in 1876 by Danish physician Julius Thomsen. In 1971, German physician Emil Peter Becker described a variant of myotonia congenita, which is characterized by more severe symptoms and a different order of inheritance. Therefore, other names for congenital myotonia are Becker's disease or Thomsen's disease.
The disease is rare. According to J. Waiton, F. Nattruss (1974), it occurs with a frequency of 0.3:100,000, according to P. Becker (1972) - 0.7:100,000. The prevalence of Thomsen and Becker myotonia in Europe is estimated as 1 : 100,000 families, in Scandinavian countries - 1:10,000 [2,9,10]. Both myotonias (Thomsen's and Becker's) are allelic variants caused by mutations in the skeletal muscle chloride channel gene CLCN1. The CLCN1 gene encodes one of the skeletal muscle chloride channels Clc1 and is located on the long arm of chromosome seven (7q35). More than 150 mutations have been described in the CLCN1 gene, most of which are missense mutations [3,4,8].
Myotonia can be inherited in an autosomal dominant manner (Thomsen's disease) and in an autosomal recessive manner (Becker's disease). These forms of the disease have a similar clinical picture, but Becker myotonia is characterized by early manifestation and more severe clinical manifestations [6].
Pathological impairment of muscle relaxation during myotonia is caused by a decrease in the permeability of the plasmalemma of muscle fibers for chlorine ions, a shift in ionic equilibrium and a violation of mediator metabolism. This leads to disruption of the contractile function of myofibrils and hyperexcitability of muscle fibers, therefore, to difficulty in muscle relaxation after contraction. This causes muscle hypertrophy to be most pronounced in men, since their muscle mass is more developed than that of women. Therefore, patients with myotonia often look well athletic and successfully perform static strength exercises [1,7].
The minimum diagnostic features are clinical and electromyographic signs of myotonia [5].
Myotonia is detected by percussion of skeletal muscles in the form of short (lasting several seconds) twitching or muscle spasms. Most often, only the lower limbs are involved in the process, sometimes the muscles of the eyeballs and hands. The most typical type is generalized, painless stiffness, which is aggravated by rest and relieved by work. The movements are slow and become faster when repeated. Typically, diffuse hypertrophy of the muscles of the thigh, forearm, and shoulder is observed, which can spread to the masticatory muscles and neck muscles.
The teenager, born in 1999, played ice hockey at a children's youth sports school for 7 years. Training during the week was 6 times from 1 hour 30 minutes to 2 hours a day.
During an annual medical examination in the State Autonomous Institution of the Republican Medical and Physical Education Dispensary in 2013, a neurologist identified clinical and anamnestic data of myotonia (at the age of 14, and at the 6th year of playing sports).
Upon examination by a neurologist, the athlete complained of muscle tension in the upper and lower extremities due to physical activity, as well as the inability to open his hands. There is a change in the voice in the form of a “grinding”.
Objectively, hypertrophy of the muscles of the upper and lower extremities was noted. On palpation the muscles are dense. Percussion of the myotonic muscles caused the formation of a prolonged myotonic ridge. Tendon reflexes are normal. Muscle strength 5 points.
For the purpose of consultation, he was sent to a medical genetic consultation (Republican Perinatal Center) for the purpose of conducting needle electromyography (EMG). On needle EMG: m. tib. ant. — spontaneous activity of the PF, POV in the form of myotonic discharges; m. ext. dig. brevis - spontaneous activity of the PF, POV in the form of myotonic discharges. The obtained needle EMG data are consistent with Thomsen's myotonia.
Taking into account the anamnesis, physical and instrumental data, the geneticist made a diagnosis: Thomsen/Becker myotonia, a moderate myotonic phenomenon. AR? or blood pressure? (no clinical symptoms were identified in the teenager’s parents).
According to an in-depth medical examination, an atrial rhythm of 59 beats/min and bradycardia were noted on the electrocardiogram with exercise. Incomplete blockade of the right bundle branch. After exercise - atrial rhythm with heart rate 74 beats/min.
According to the results of echocardiography, an additional chord of the left ventricle, degree 1 mitral valve prolapse (up to 0.41 cm), and hemodynamically insignificant aortic regurgitation were revealed. EF 68%, SV 73%.
According to the literature, this case of Thomsen/Becker myotonia in a teenager involved in sports is typical.
Table 1
Signs of Thomsen/Becker myotonia in a teenager involved in sports
| № | Signs | Literary data | Clinical case |
| 1 | Debut | Children's age 10–12 years, less often at a later age. | 14 years old. |
| 2 | Complaints | Local increase in muscle tone (muscles of mastication, hands of the upper extremities). | Tension of the muscles of the upper and lower extremities due to physical activity, slower relaxation of muscles in the hands. |
| 3 | Clinical picture | Voice change. | Change in voice - “grinding”. |
| 4 | Objectively | Signs of increased mechanical excitability of muscles. | Hypertrophy of the muscles of the upper and lower extremities, muscles are compacted, myotonic roll. |
| 5 | Patient's appearance | Athletic build. | Athletic build. |
| 6 | Clinical manifestations | Cold, physical activity. | Exercise stress. |
| 7 | Cardiac pathology | Left ventricular hypertrophy, arrhythmia. | Mitral valve prolapse of the first degree (up to 0.41 cm), aortic regurgitation is hemodynamically insignificant. |
| 8 | Instrumental data - needle EMG | Myotonic discharges. | Myotonic discharges. |
Some signs of the disease in a teenager involved in sports can be attributed to the manifestation of training, such as an athletic physique and hypertrophy of the muscles of the upper and lower extremities. But with a thorough objective examination and instrumental data, Thomsen/Becker myotonia is confirmed.
In our opinion, greater physical activity could worsen the clinical manifestations of myotonia, but during an annual in-depth medical examination of athletes, the disease was detected with minimal manifestations.
After the diagnosis was made by a geneticist, observation by a neurologist at the place of residence and symptomatic treatment were prescribed. The teenager's condition is satisfactory.
Taking into account objective data, the absence of pronounced clinical manifestations and the observation of a neurologist, a sports medicine doctor, as well as the fact that myotonia decreases with age and muscle strength does not decrease, the teenager moved from the ice hockey sports school to the general physical training group in the sports section. Thai boxing Classes are held 3 times a week, 1 hour a day. Training is carried out according to an individual plan, under the supervision of muscle spasms of the upper and lower extremities.
Literature:
1. Badalyan L. O. Child neurology. Textbook 3rd ed., revised. and additional - M.: Medicine, 1984. - 576 p.
2. Ivanova E. A., Polyakova A. V. Population frequency and reasons for the prevalence in the Russian population of the mutation p.ARG894* in the CLCN1 gene, which controls the development of Thomsen and Becker myotonia // Genetics, 2013, volume 49, No. 9, P. 1–9.
3. Ivanova E. A., Fedotov V. P., Dadali E. L., Rudenskaya G. E., Kurbatov S. A., Polyakov A. V. Molecular genetic analysis of the causes of non-dystrophic myotonia // Material of the VIth Congress of the Russian Society of Medical Genetics, Medical Genetics, supplement to No. 5, 2010, P.72.
4. Ivanova E. A., Fedotov V. P., Kurbatov S. A., Polyakov A. V. Molecular genetic analysis of the causes of hereditary Thomsen-Becker myotonia // Medical Genetics, Vol. 8, No. 12(90), 2009 g., p.38.
5. Kozlova S. I., Demikova N. S., Semanova E., Blinnikova O. E. Hereditary syndromes and medical genetic counseling. Atlas-directory. Ed. 2nd add. - M.: Praktika, 1996. - 416 pp. P. 163.
6. Kurbatov S. A., Fedotov V. P., Ivanova E. A., Galeva N. M. Phenogenetic correlations of hereditary myotonic syndromes // Materials of the VI Congress of the Russian Society of Medical Genetics, Medical Genetics, Appendix No. 5, 2010, p.99.
7. Rudenskaya G. E., Ivanova E. A., Galeva N. M. A case of Becker’s myotonia with equine foot deformity // Journal of Neurology and Psychiatry named after. S. S. Korsakova. 2012.-No. 8.- P. 70–71.
8. Ivanova E., Fedotov V., Dadali E., Rudenskaya G., Kurbatov S., Myotonia congenital (MC) in Russia: the two frequent mutations in CLCN1gene allows to reveal 14.0% mutant chromosomes of patients with MC. //European journal of Human Genetic, Volume 18 Supplement 1 June 2010. P. 87–88.
9. Koch M., Harley H., Sarfarazi M. et al. Myotonia congenital (Thomsen's disease) excluded from the region of the myotonic dystrophy locus on chromosome 19. - Hum. Genet., 1989, v. 82, p. 163–166.
10. Torbergsen T. A family with dominant hereditary myotonia, muscular hypertrophy and increased muscular irritability, distinct from myotonia congenita Thomsen. — Acta Neurol. Scand., 1975, v. 51, p. 225–232.
Differential diagnosis of Rossolimo-Steinert-Kurshman myotonia
It is necessary to differentiate Rossolimo-Steinert-Kurshman myotonia from other known types of myotonia, which in some situations may have similar signs and course. Atrophy of muscle tissue is characteristic exclusively of this disease and allows you to quickly distinguish it from Thomsen's myotonia, a sign of which is muscle hypertrophy. Early involvement of the facial muscles and dominant inheritance distinguish it from Becker myotonia. In addition to these conditions, it is necessary to differentiate Rossolimo-Steinert-Kurshman disease from ALS and Charcot-Marie-Tooth amyotrophy.
Treatment of Rossolimo-Steinert-Kurshman myotonia
In modern medical practice, there is currently no therapy designed to completely cure people of Rossolimo-Steinert-Curschmann myotonia. Patients diagnosed with this disease are prescribed a diet based on foods with minimal or no potassium levels. People suffering from this myotonia should not be overcooled, since low temperatures can provoke spasms at any time.
To reduce the risk of their sudden appearance and improve the general condition of the patient, he is prescribed the following drugs: quinine, novocainamide, diphenine simultaneously with diacarb. To these medications, experts recommend adding the mandatory use of anabolic steroids: nerobol, retabolil, methylandrostenediol. It is also necessary to include small doses of ATP and B vitamins in the list of required substances.
The above drugs have a good effect in both the classical and congenital types of the disease. All of these measures can reduce the negative effects of the disease on the patient’s body and prolong his life, but they are in no way able to completely rid him of myotonia and make him an absolutely healthy person again.