Medical information is reliable Checked by Shaidullin Renat Flyurovich
Creutzfeldt-Jakob disease is a rare type of degenerative brain disease. It is part of a group of prion transmissible encephalopathies, which are caused by the entry of an abnormal isoform of the prion protein into the body and its accumulation in neurons.
The pathology occurs in one in a million people on the globe. The disease was first described in 1920 by the German neurologist Hans Creutzfeldt. And a year later, his fellow countryman and colleague Alfons Jacobs clarified that the deviation is characterized by the manifestation of mental disorders against the background of organic damage to the pyramidal, extrapyramidal region of the central nervous system, the anterior part of the horns of the spinal cord, the cerebellum and visual impairment associated with changes in cortical structures.
The incidence of this pathology is low - it is registered in one person per million. In the 90s of the last century, symptoms of a new variant of Creutzfeldt-Jakob disease began to be noted, which were of an infectious nature and were associated with infection from cows and other cattle (“mad cow disease”). Most often, the disease is registered in people over the age of 65, but in recent years, deviations of this type have also been observed in younger patients.
What causes Creutzfeldt-Jakob syndrome
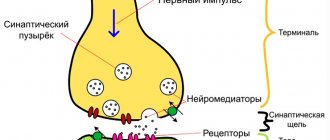
The disease is caused by the presence in the body of a special protein (prion), which has a disrupted structure and has a negative effect on the nervous system. The action of a prion is reminiscent of the effect of viruses that cause cell death during their reproduction, but it does not carry genetic material (DNA or RNA).
In its desire to get rid of the aggressor, the cell that is being attacked begins to actively release substances. But the protein deposited on the membrane does not allow them to come out, and they destroy the organelles and trigger the process of apoptosis. Neighboring structures begin to react to this with inflammation, and therefore further cell damage occurs.
Prions can withstand a wide temperature range, radiation, and are resistant to enzymatic degradation. They are not affected by antibiotics and antivirals. Under the influence of this protein, the tissue of the nervous system becomes “leaky” and begins to resemble a sponge.
Exogenous infection (in addition to spontaneous internal transformation of prions and the hereditary form of the disease) occurs from the external environment in the following ways:
- When eating food of animal origin. Especially a lot of the infectious agent “mad cow disease” is observed in beef, brain, blood and spleen. Cases of the disease are described among peoples who practice ritual cannibalism.
- When transplanting internal organs, including liver, kidneys, cornea, heart muscle. The possibility of transmission remains during transfusion of both whole blood and its individual components (mass of red blood cells, leukocytes, plasma).
- The use of drugs obtained from animal tissue.
- Transmission from person to person through biological fluids by getting them onto wound surfaces or into the stomach.
Creutzfeldt-Jakob disease is not transmitted by airborne droplets, as is the case with viral infections, or by ordinary touch. There are suggestions of transmission of the pathological prion protein through sputum, urine and feces, but there has been insufficient research into this variant of infection.
Pathogenesis of the disease
Currently, there are two types of pathogenesis of Creutzfeldt-Jakob disease. The first is extracerebral, which does not relate to brain structures and cerebral. Both types occur when infected with prions from the outside, and the hereditary and sporadic forms of the disease are still insufficiently studied.
The extracerebral variant of the development of the pathology is that the distorted protein enters the body and combines with lymphocytes, macrophages and other cells involved in immune defense. It is transported by them to the tissues of the lymphatic system (spleen, lymph nodes). There it multiplies, and the body does not interfere with this, since it does not perceive the prion as a foreign body. At the same time, it does not damage or cause enlargement of lymphoid organs, but only multiply and persist in them.
Subsequent spread of the pathogen occurs under the influence of the autonomic nervous system. They are sent to the central nervous system, and the cerebral stage of Creutzfeldt-Jakob disease begins. They accumulate in intracellular fluid and nerve endings. The result is a decrease in normal prion protein, which is involved in normal cell nutrition.
2. Reasons
It should be noted that today there are four main forms of the disease, and this classification is based on the etiological (causal) criterion.
The classic form of Creutzfeldt-Jakob disease is caused by the spontaneous occurrence of prions in the body; it usually occurs in old age and occurs with an incidence of only a few cases per million population.
The low but statistically significant probability of repeated cases of the disease within the same family forces us to identify a hereditary form of prion transmission (transmission).
The iatrogenic form involves the accidental introduction of prion material during surgical operations and other medical procedures and manipulations.
Finally, since 1995, the so-called “new variant”, which is registered mainly in young people in the UK and, according to reasoned assumptions of experts, is a direct consequence of the consumption of beef containing prions.
Visit our Neurology page
Classification of Creutzfeldt-Jakob disease
Depending on the characteristics of the symptoms, the following forms are distinguished in Creutzfeldt-Jakob disease:
- subacute encephalopathy with diffuse tissue damage and rapid progression;
- dyskinetic, or classic (a combination of extrapyramidal and pyramidal disorders with manifestations of dementia);
- intermediate with a predominance of symptoms of damage to the cerebellum and subcortical areas;
- amyotrophic (with speech and movement disorders).
According to etiology, Creutzfeldt-Jakob disease is divided into the following types:
- Spontaneous. The second name is classic. In this case, prions do not enter the body from the outside, but are formed for no apparent reason. Most likely, modern medical science simply cannot find an explanation for this phenomenon. Older people (after 50 years) are more often affected.
- Hereditary. With such a deviation, the formation of a prion is genetically determined.
- Iatrogenic. In this type of disease, the damaging protein enters the body from the outside. Previously, some drugs could serve as its source. Most of them are currently not in use. The cause is also the membranes taken from the tissue of corpses to close wounds during brain surgery.
- A new form of Creutzfeldt-Jakob disease. First described by UK doctors in 1995. The reason for this was the consumption of meat from “mad cows” containing prions. Unlike the classic version, the pathology is observed in people over the age of 20 and manifests itself in the form of personality changes.
Creutzfeldt-Jakob disease
prion proteins are not similar in amino acid sequence to yeast prion proteins. Despite this, the main structural features (formation of amyloid fibers and high specificity that prevents the transfer of prions from one type of organism to another) are common to them. However, the prion responsible for mad cow disease has the ability to be transmitted from species to species. Studies of the brain tissue of animals that died from prion infections showed that prions do not contain nucleic acids, but are proteins. the first characterized prion proteins was PrP (from English prion-related protein or proteinase - resistant protein ) with a mass of about 35 kDa. It is known that PrP can exist in two conformations - “healthy” - PrPC, which it has in normal cells (C - from the English cellular - “cellular”), in which alpha helices predominate, and “pathological” - PrPSc, actually prion (Sc- from scrapie), which is characterized by the presence of a large number of beta strands. When entering a healthy cell, PrPSc catalyzes the transition of cellular PrPC to the prion conformation. The accumulation of prion protein is accompanied by its aggregation, the formation of highly ordered fibrils (amyloids), which ultimately leads to cell death. The released prion appears to be able to penetrate neighboring cells, also causing their death. The functions of the PrPC protein in a healthy cell have not yet been determined. Normally, the PrPC protein is associated with the cell membrane and is glycosylated with a sialic acid residue. It makes cyclic transitions into the cell and back to the surface during endo- and exocytosis. One such cycle lasts about an hour. In an endocytic vesicle or on the cell surface, the PrPC molecule can be cut into two approximately equal parts by proteases. The mechanism of spontaneous occurrence of prion infections is not completely clear. It is believed (but not yet fully proven) that prions are formed as a result of errors in protein biosynthesis. Mutations of genes encoding prion protein (PrP), translation errors, proteolysis processes are considered the main candidates for the mechanism of prion formation. According to recent research, prions are capable of Darwinian evolution through the action of natural selection. There is evidence to suggest that prions are not only infectious agents, but also have functions in normal bioprocesses. For example, there is a hypothesis that the mechanism of genetically determined stochastic aging occurs through prions. A person can become infected with prions contained in food, since they are not destroyed by enzymes in the digestive tract. Freely penetrating through the wall of the small intestine, they ultimately enter the central nervous system. This is how a new variant of Creutzfeldt-Jakob disease (nvCJD) is transmitted, which people become infected with after eating beef containing nerve tissue from cattle with bovine spongiform encephalopathy (BSE, mad cow disease). Prions can also enter the body parenterally. Cases of infection have been described through intramuscular administration of drugs made from human pituitary glands (mainly growth hormones for the treatment of dwarfism), as well as infection of the brain with instruments during neurosurgical operations, since prions are resistant to currently used thermal and chemical methods of sterilization. This form of Creutzfeldt-Jakob disease is designated iatrogenic (1CJD). Under certain, unknown conditions, spontaneous transformation of a prion protein into a prion can occur in the human body. This is how the so-called sporadic Creutzfeldt-Jakob disease (sCJD) arises, first described in 1920 independently by Hans Gerhard Creutzfeldt and Alphonse Maria Jakob. It is believed that the spontaneous occurrence of this disease is due to the fact that normally a small number of prions are constantly generated in the human body, which are effectively eliminated by the cellular Golgi apparatus. Violation of this ability of “self-cleaning” of cells can lead to an increase in the level of prions above the permissible normal limit and to their further uncontrolled spread. According to this theory, the cause of sporadic Creutzfeldt-Jakob disease is a dysfunction of the Golgi apparatus in cells. A special group of prion diseases are hereditary (congenital) diseases caused by a mutation in the prion protein gene, which makes the resulting prion protein more susceptible to spontaneous changes in spatial configuration and their transformation into prions. This group of hereditary diseases also includes the hereditary form of Creutzfeldt-Jakob disease (fCJD), which is observed in a number of countries around the world. In prion pathology, the highest concentration of prions is found in the nervous tissue of infected people. A significant number of prions are found in lymphatic tissue. The presence of prions in biological fluids, including saliva, has not yet been unequivocally confirmed. If the idea of a constant occurrence of small numbers of prions is correct, then it can be assumed that new, more sensitive diagnostic methods will discover these numbers of prions scattered throughout various tissues. In this case, however, we will talk about the “physiological” level of prions, which do not pose any threat to humans.
Creutzfeldt-Jakob disease: symptoms
Creutzfeldt-Jakob disease has characteristic symptoms, regardless of origin. Iatrogenic, hereditary and classical forms manifest themselves in approximately the same way. The new type of disease proceeds somewhat differently.
Main manifestations
The initial stage is manifested by temporary loss of memory of events, changes in mood, and loss of interest in others. Gradually, the patient experiences more and more difficulties in carrying out vital actions. The final stage is accompanied by blurred vision, slow speech and the appearance of hallucinations.
In approximately 40% of patients with the sporadic form, the disease progresses subacutely, with gradually worsening cognitive impairment. Another 40% of those infected suffer from dysfunction of the cerebellum, and the rest have a mixed form of the disease.
The most characteristic clinical symptoms of classical Creutzfeldt-Jakob disease are:
- conduct disorder;
- memory impairment;
- diplopia and development of cortical blindness;
- mental disorders;
- cerebellar dysfunction, ataxia;
- lack of coordination;
- paresis;
- dysarthria;
- inability to self-care, sloppiness and other signs of dementia;
- a combination of signs of damage to the pyramidal and extrapyramidal systems.
Many patients experience local or generalized types of epileptic seizures. They appear in response to certain stimuli - light, sound, touch.
The terminal stage is accompanied by global cognitive impairment and complete inability to move. Death usually occurs within 8 months to 2 years after the onset of the initial clear symptoms of the disease. The new form of the disease is characterized by mental disorders and changes in sensitivity that come to the fore.
Stages
Experts distinguish three stages of Creutzfeldt-Jakob disease:
- Prodromal stage. Its symptoms do not have specific manifestations and are observed in approximately 30% of all patients. The person experiences asthenia, sleep and appetite disturbances, decreased memory ability, weight loss, and mild cognitive impairment. The patient is characterized by the appearance of phobias, delusions, and hallucinations.
- Initial stage. Changes in vision begin, headache appears, instability, ataxia, sensory disturbances, tremor, myoclonus, epileptic seizures, and other neurological symptoms increase against the background of dementia.
- The advanced stage is accompanied by spastic paralysis, all signs of disturbances in the functioning of brain structures, and a severe form of dementia without the possibility of contact with the patient. A person completely loses control over the functioning of the pelvic organs, swallowing is impaired, and a decrease in temperature is noted.
Most often, Creutzfeldt-Jakob disease develops gradually, but sometimes it has an acute and subacute onset. Death occurs as a result of failure of vital centers and severe exhaustion.
Diagnosis of Creutzfeldt-Jakob disease
A presumptive diagnosis of Creutzfeldt-Jakob disease is made based on clinical manifestations and additional research methods. Pathology can be suspected if there are a combination of certain symptoms observed over a long period of time:
- progression of dementia;
- pyramidal and extrapyramidal disorders;
- myoclonus;
- disorders of the cerebellar structures;
- visual impairment.
To clarify the pathology, the neurologist refers the person to undergo encephalography, MRI of the brain, spinal puncture, stereotactic biopsy:
- On the EEG, almost every patient with Creutzfeldt-Jakob disease shows a decrease in electrical activity with periodic sharp waves. Changes characteristic of myoclonus are observed in 50% of patients. A new type of disease often does not manifest itself as any abnormalities on the encephalogram.
- MRI can reveal signs of atrophic changes in the cerebellum and cerebral cortex (honeycomb syndrome), and dilatation of the ventricles. Areas with increased signal are sometimes detected from subcortical structures and the thalamus.
- Lumbar puncture is mandatory if Creutzfeldt-Jakob disease is suspected. The study of cerebrospinal fluid allows for a differential diagnosis with other pathological disorders of the central nervous system.
- The most objective way to reliably determine the diagnosis is a morphological examination of a biopsy of brain tissue. Using the immunochromatographic method, it is possible to detect the prion protein. Its presence suggests that the patient has Creutzfeldt-Jakob disease.
Differential diagnosis must be made with other diseases of the central nervous system that occur with similar symptoms:
- senile, multi-infarct dementia;
- Alzheimer's disease;
- Hashimoto's encephalopathy;
- inflammatory damage to the meninges (encephalitis, arachnoiditis, meningitis).
Diagnostics
Diagnosis of Creutzfeldt-Jakob disease is based on data from the clinical picture, neurological status, and cases of consumption of animal meat from outbreaks of animal spongy encephalopathy.
It is also necessary to exclude other pathologies that could lead to clinical manifestations of the disease. Thus, an EEG study reveals background plane oscillations in the form of waves consisting of three phases. These waves can be superimposed by sharp and slow high-amplitude waves with a frequency of about 2 Hz.
MSCT and MRI studies reveal signs of brain tissue degeneration. Blood and cerebrospinal fluid, as a rule, do not change. A reliable diagnosis is considered after a lifetime brain biopsy using special molecular research methods. If it is impossible to conduct the study, or if the patient or his legal representatives disagree, the diagnosis is considered probable, but not confirmed.
Treatment of Creutzfeldt-Jakob disease in Moscow
Etiotropic therapy for the disease is still under development. Therefore, all treatment in the clinic is aimed at relieving symptomatic manifestations. All previously used medications are discontinued for the patient, since they can negatively affect the course of the disease, exacerbating the disturbance of mnestic functions and worsening the patient’s behavior.
Conservative therapy
It has already been proven that traditional means that are successfully used in the treatment of viral infections (vaccines, interferons) do not have a positive effect on the course of Creutzfeldt-Jakob disease. Some results with a temporary improvement in condition can be obtained by introducing an antibiotic, which blocks the work of the Golgi apparatus inside the cell and prevents the proliferation of prions. It has been noted that when using certain calcium channel blockers, the duration of functioning of the affected brain structures increases over time.
A slight slowdown in the accumulation of pathological proteins is observed when using an immunomodulator, which stimulates the production of endogenous interferon. But these drugs have strong side effects.
Symptomatic therapy in patients is aimed at eliminating:
- tremors and epileptic seizures;
- extrapyramidal disorders;
- depression and anxiety;
- pain syndrome.
For this purpose, various groups of drugs are used, their choice is made by the doctor. The basis for the use of a particular remedy is the presence and severity of the deviation. The following groups of drugs are used in therapy:
- neuroleptics;
- tranquilizers;
- analgesics;
- opiates;
- sedatives;
- antidepressants;
- anticonvulsant medications.
The advantage of having the patient in the clinic
After a diagnosis of Creutzfeldt-Jakob disease is made, a person needs to provide full symptomatic therapy, psychological support for the patient and his family members. The further the pathology progresses, the more care and attention the patient needs, as he gradually loses self-care skills and the functioning of the pelvic organs is disrupted.
Working relatives cannot provide round-the-clock vigil near the patient. And he will need to be fed, washed and helped with movement as needed. In some cases, he has to remove urine through catheterization as a result of bladder dysfunction. And this manipulation can only be performed by specialists with medical education.
If swallowing function is impaired, a person will need nutrition using a special tube and drip solutions with the necessary substances. At home, it is possible to provide full treatment and care only in the earliest stages. Subsequently, the patient will definitely require systematic care from specially trained personnel.
In the terminal stage, measures are taken to prolong life. They are carried out only in the presence of resuscitation equipment - mechanical ventilation, oxygen therapy, means for stimulating the respiratory and cardiovascular systems.
When choosing a clinic to provide care to patients with Creutzfeldt-Jakob disease, relatives should understand that the highest quality care can be provided in an institution that employs experienced specialists with a specific profile.
If you hospitalize a person in a public hospital, you can save money, but the conditions of stay will not be ideal:
- several people are accommodated in the wards;
- you will come to purchase medications on your own, since those that are available cannot always effectively eliminate complications;
- the patient is at high risk of contracting a hospital-acquired infection;
- staff will not be able to constantly monitor the patient and provide assistance if necessary 24 hours a day.
When the patient is still conscious and able to assess the current situation, his stay in a government institution will significantly aggravate the condition, cause depression and increased mental disorders.
CLINICAL CASE STUDY OF CREUTZFELDT-JACOB DISEASE Tolstolutskaya A.O., Delinskaya D.A.
Text of a scientific article
Introduction: Prion diseases have a long incubation period, a malignant course and lead to rapid death. These sufferings are caused by an abnormal variety of prions. The causative agent of prion diseases is the infectious prion protein Prion Protein - scarpie (PrPSc), which is formed as a result of conformational changes in the normal (non-infectious) cellular protein Prion Protein - cellular (PrPc) [1, 58]. The accumulation of the pathological form of PrPSc leads to massive death of nerve cells. Prions are the only known infectious agents whose reproduction occurs without the participation of nucleic acids. The question of whether prions should be considered a form of life is currently open. Creutzfeldt-Jakob disease (CJD) is one of four known rare human prion diseases. This disease was described in the 1920s by G. G. Creutzfeldt and A. M. Jacob. CJD is rightfully one of the most dangerous infectious diseases, even despite its extremely rare prevalence: 0.5-1.5 cases per 1 million population. 90% of all cases of CJD occur in the classical form; men are more often affected [2, 68]. The total number of registered cases of the CJD variant in 7 European countries and 4 other countries was 219 observations [3, 599]. We unified the existing classifications into a single one, dividing them into endogenous (sporadic, hereditary and new) and exogenous (iatrogenic) forms. The leading symptoms during the observation process in patients were: increasing intellectual-mnestic disorders, amnesia, fear, depression and dementia, dysarthria and dysphonia, ataxia, extrapyramidal rigidity, tetraparesis, amyotrophy, ophthalmoparesis, convulsive syndrome, myoclonus, trembling of the head and limbs, insomnia, hyperthermia [4]. The International CJD Consortium has proposed specific criteria for diagnosing CJD. These criteria take into account: I. Clinical symptoms: • Dementia. • Cerebellar or visual. • Pyramidal or extrapyramidal. • Akinetic mutism. II. Data from additional studies: • Typical wave complex (PSWC phenomenon) on the EEG. • Determination of protein 14-3-3 in the CSF (in patients with disease duration less than 2 years). • Increased signal intensity from the caudate nucleus and putamen or from at least 2 cortical regions (temporal-parietal-occipital) in DWI or FLAIR mode. According to this scheme, to make a probable diagnosis of CJD, two criteria from point I and at least one criterion from point II must be present [5, 64]. The greatest danger in terms of the development of the epidemic is the iatrogenic form. In 1974, the first case of CJD developing in a patient was reported 18 months after a corneal transplant from a donor with established CJD. Proven methods of transmission: transplantation of donor infected cornea and dura mater, treatment with drugs of human growth hormone and gonadotropins, use of infected instruments and intracerebral electrodes during neurosurgical operations. Isolated cases of CJD have been described among neurosurgeons, pathologists and veterinary workers. The incubation period ranged from 4 to 30 years [6, 2661]. We described a case of CJD in a patient with a humerus transplant at the age of 18. Clinical case: Patient L., 31 years old, was admitted to the neurology clinic of Rostov State Medical University with severe mental and behavioral disorders. I made no complaints. According to relatives, neurological symptoms appeared in July 2021. When contacting a doctor, the patient's condition was regarded as cerebroasthenic syndrome. Therapy was prescribed without a significant positive effect. In August 2021, magnetic resonance imaging (MRI) was performed: an MR picture of single local arachnoid changes of a liquor cystic nature, no convincing MR data for the presence of focal changes in the brain substance were identified. In September 2021, violent movements in the hands, sleep disturbances, speech disorders in the form of echolalia began, the patient found it difficult to find words, he became untidy, and forgot about eating. The gait was severely impaired, such as apraxia of walking. According to relatives, during September 2021, he lost self-care skills, lost speech contact, and could not complete tasks on command or by imitation. The patient was consulted by a psychiatrist and diagnosed with dissociative disorder of the pseudodementia type. From the life history: humerus transplantation in 1996. A number of studies have been conducted: Electroencephalography (EEG): A flattened type of EEG. Significant diffuse bioelectrical activity of the brain of a regulatory nature. Decreased functional lability of the cortex. Typical epileptiform activity was not recorded. MRI of the brain: MR signs of moderate dilatation of the subarachnoid spaces. Magnetic resonance angiography (MRA) did not reveal any pathology of the vessels of the arterial circle. Biochemical blood test: total bilirubin (39.2 µmol/l), urea (7.7 mmol/l) are increased. Ultrasound examination (ultrasound) of the kidneys: signs of moderate diffuse changes in the parenchyma and pyelocaliceal system (PCS). Ceruloplasmin: 0.218 g/l (N = 0.2-0.6 g/l). Consultation with an ophthalmologist: Angiopathy of the retinal vessels of both eyes, there is no evidence of the presence of Kayser-Fleshner rings on the cornea, retinal edema, or congestive optic discs. Ultrasound of the abdominal organs: minor diffuse changes in the pancreatic parenchyma. Vague signs of cholecystopancreatitis. Gastroenterologist: hepatocerebral degeneration cannot be excluded. A biochemical blood test (alanine aminotransferase (ALT), aspartate aminotransferase (AST), alkaline phosphatase, bilirubin, urea) over time is recommended. I was admitted to the neurology clinic to clarify the diagnosis. Neurological status: not oriented in place, time, self. Not available for productive verbal contact. When testing higher brain functions, severe sensory aphasia and motor apraxia were revealed, so diagnosing other cortical disorders was not possible. Assessing phonation and articulation function is difficult. It is impossible to check the pharyngeal reflexes due to the patient’s active resistance; the tongue does not show itself, and when trying to stick it out, it quickly hides it back. Reflexes of oral automatism (palmomental, proboscis) are evoked. The range of active movements in the joints of the upper extremities is reduced. The phenomenon of paratonia. Cortical myoclonus. Muscle strength in the limbs up to 5 points. Grasping automatisms. Passive movements: muscle tone is increased according to the extrapyramidal type, more on the right. Tendon reflexes from the upper and lower extremities are high D>S, with expansion of reflexogenic zones, without pathological foot signs. It is not possible to assess sensory impairments; in the Romberg position, he is not stable with his eyes closed and open. He does not perform the finger-knee test and the finger-nose test because he does not understand the task. There is no nystagmus. Controls pelvic functions. Meningeal symptoms: negative. A repeat EEG was performed: a flattened curve. The reactivity of the brain systems is sharply reduced. When carrying out provoking AF on FN 3-27, there is a slowdown in bioelectrical activity in the left occipito-parietal and right anterior temporal regions. When carrying out photostimulation tests on FT-2, FT-6, FT-10, a slowdown in bioelectrical activity is observed in the left hemisphere with an emphasis on the parietal-central-frontal region. Considering the patient’s young age, short medical history, rapid development of symptoms over three months, the presence of frontal syndrome (euphoricity, uncriticality of one’s condition, foolishness), lesions of the extrapyramidal system (increased muscle tone of a plastic type), severe cognitive impairment (pseudo-dementia), anamnestic data (humeral bone transplant in 1996), exclusion of the AIDS-dementia complex and EEG data (flattening of the curve and absence of epileptic complexes), a diagnosis was suggested: Creutzfeldt-Jakob disease. Despite an MRI of the brain, to confirm the diagnosis, an MRI was repeated in DWI mode: MRI signs of damage to the cerebral cortex and subcortical nuclei of the brain on the left. Changes characteristic of spongiform encephalopathy (Creutzfeldt-Jakob disease). Conclusions: • Lack of alertness among practicing physicians regarding rare fatal dangerous infectious diseases in the presence of rapidly developing dementia in young and middle age. • The feasibility of performing MRI in DWI and FLAIR modes to confirm the diagnosis of CJD. • Possibility of infection of patients from cadaveric bone material. • The need to develop a clear treatment plan after diagnosis.
Questions and answers
If you are in constant contact with a person who has Creutzfeldt-Jakob disease, is there a chance of becoming infected?
Infection through normal contact is extremely rare. But during care you should adhere to hygiene rules. They include washing your hands before eating, protecting your skin from body fluids, and covering all wound surfaces with waterproof bandages.
Is it possible to cure a person with this disease?
This disease cannot be treated and is fatal with a 100% probability. All that can be done in this case is to provide decent care for the patient, stop emerging complications and carry out symptomatic therapy in order to improve the quality of life.
Why does Creutzfeldt-Jakob disease cause death?
As the pathology progresses, various complications develop, which most often become the cause of death. Some of them are amenable to drug correction - epileptic seizures, acute psychoses, pneumonia due to infection. Proper care can reduce a person's suffering from pressure sores resulting from muscle degeneration. The final outcome of the disease is the development of respiratory or cardiovascular failure, which ends in death. But even these functions can be maintained for some time with the help of special devices.