Медицинская информация достоверна Проверял Шайдуллин Ренат Флюрович
Болезнь Крейтцфельдта-Якоба относится к редким видам дегенеративных поражений головного мозга. Она входит в группу прионных трансмиссивных энцефалопатий, которые вызываются попаданием в организм аномальной изоформы белка приона и его накапливание в нейронах.
Патология возникает у одного из миллиона человек на земном шаре. Заболевание было впервые описано в 1920 году германским невропатологом Гансом Кройцфельдтом. А спустя год его земляк и коллега Альфонс Якобс уточнил, что для отклонения характерно проявление психических нарушений на фоне органического поражения пирамидальной, экстрапирамидальной области ЦНС, передней части рогов спинного мозга, мозжечка и нарушениями зрения, связанное с изменениями корковых структур.
Частота возникновения этой патологии невелика – она регистрируется у одного человека на миллион. В 90 годах прошлого столетия стали отмечаться симптомы нового варианта болезни Крейтцфельдта-Якоба, которые имели инфекционную природу и связывались с заражением от коров и другого крупного рогатого скота («коровье бешенство»). Чаще всего заболевание регистрируется у людей в возрасте от 65 лет, но в последние годы отклонения такого типа отмечаются и у более молодых пациентов.
Что вызывает синдром Крейтцфельдта-Якоба
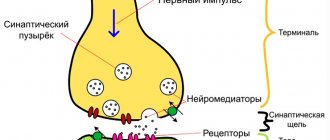
Заболевание вызывается наличием в организме особого белка (приона), который имеет нарушенную структуру и оказывает негативное влияние на нервную систему. Действие приона напоминает эффект вирусов, вызывающих гибель клетки при своем размножении, но при этом он не несет в себе генетический материал (ДНК или РНК).
В своем стремлении избавиться от агрессора, клетка, которая подвергается атаке, начинает активно выделять вещества. Но белок, осевший на мембране, не позволяет им выйти наружу, и они разрушают органеллы и запускают процесс апоптоза. Соседние структуры начинают реагировать на это воспалением, в связи с чем и происходит дальнейшее поражение клеток.
Прионы выдерживают большой температурный диапазон, облучение, они устойчивы к ферментативному расщеплению. На них не оказывают влияния антибиотики и противовирусные средства. Под воздействием этого белка ткань нервной системы становится «дырявой» и начинает напоминать губку.
Заражение экзогенным путем (помимо спонтанной внутренней трансформации прионов и наследственной формы заболевания) происходит из внешней среды такими способами:
- При употреблении пищи животного происхождения. Особенно много инфекционного агента «коровьего бешенства» наблюдается в говядине, мозге, крови и селезенке. Описываются случаи заболевания у народов, которые практикуют ритуальный каннибализм.
- При пересадке внутренних органов, в том числе печени, почек, роговицы, сердечной мышцы. Возможность передачи сохраняется при переливании как цельной крови, так и ее отдельных компонентов (массы эритроцитов, лейкоцитов, плазмы).
- Применение препаратов, полученных их ткани животных.
- Передача от человека к человеку через биологические жидкости путем попадания их на раневые поверхности или в желудок.
Болезнь Крейтцфельдта-Якоба не передается воздушно-капельным путем, как это бывает при вирусных инфекциях, а также обычным прикосновением. Есть предположения передачи патологического белка приона через мокроту, мочу и кал, но исследований этого варианта заражение проводилось недостаточно.
Патогенез болезни
В настоящее время выделяется два типа патогенеза болезни Крейтцфельда – Якоба. Первый – экстрацеребральный, который не относится к мозговым структурам и церебральный. Оба типа возникают при заражении прионами извне, а наследственная форма заболевания и спорадическая до сих пор изучены недостаточно.
Внецеребральный вариант развития патологии заключается в том, что извращенный белок поступает в организм и соединяется с лимфоцитами, макрофагами и другими клетками, участвующими в иммунной защите. Он переносится ими в ткани лимфатической системы (селезенку, лимфоузлы). Там происходит его размножение, причем организм не препятствует этому, так как не воспринимает прион как чужеродное тело. При этом он не повреждает и не вызывает увеличения лимфоидных органов, а только размножаются и сохраняются в них.
В дальнейшем распространение патогена происходит под влиянием вегетативной нервной системы. Они направляются в центральную нервную систему, и начинается церебральный этап болезни Крейтцфельда – Якоба. Они накапливаются во внутриклеточной жидкости и нервных окончаниях. В результате происходит снижение нормального прионного белка, который участвует в обычном питании клеток.
2.Причины
Следует отметить, что на сегодняшний день выделяют четыре основные формы заболевания, и в основе такой классификации лежит этиологический (причинный) критерий.
Классическая форма болезни Крейтцфельдта-Якоба обусловлена спонтанным возникновением прионов в организме; обычно это происходит в пожилом возрасте и встречается с частотой всего несколько случаев на миллион населения.
Невысокая, но статистически значимая вероятность повторных случаев заболевания в рамках одной семьи заставляет выделять наследственную форму прионной трансмиссии (передачи).
Ятрогенная форма предполагает случайный занос прионного материала во время хирургических операций и прочих процедур и манипуляций медицинского характера.
Наконец, с 1995 года отдельно рассматривают т.н. «новый вариант», который регистрируется, в основном, у молодых лиц в Великобритании и, по аргументированным предположениям специалистов, является прямым следствием потребления содержащей прионы говядины.
Посетите нашу страницу Неврология
Классификация болезни Крейтцфельдта-Якоба
В зависимости от особенностей симптоматики, при болезни Крейтцфельдта-Якоба различают следующие формы:
- подострая энцефалопатия с диффузным поражением ткани и быстрым течением;
- дискинетическая, или классическая (сочетание экстрапирамидальных и пирамидальных нарушений с проявлениями деменции);
- промежуточная с преобладанием симптомов поражения мозжечка и областей подкорки;
- амиотрофическая (с нарушением речи и движений).
По этиологии болезнь Крейтцфельдта-Якоба подразделяется на следующие виды:
- Спонтанная. Второе название – классическая. Прионы в этом случае не попадают в организм извне, а формируются без видимой причины. Скорее всего, современная медицинская наука просто не может найти объяснением этому явлению. Чаще страдают люди старшего возраста (после 50 лет).
- Наследственная. При таком отклонении формирование приона обусловлено генетически.
- Ятрогенная. Повреждающий белок при данном виде болезни попадает в организм извне. Его источником ранее могли служить некоторые препараты. Большая часть из них в настоящее время не используются. Причиной также выступают оболочки, взятые из ткани трупов для закрытия ран при операциях на мозге.
- Новая форма болезни Крейтцфельдта-Якоба. Впервые описана медиками Великобритании в 1995 году. Причиной ее стало употребление мяса «бешеных коров» с наличием прионов. В отличие от классического варианта патология наблюдается людей в возрасте от 20 лет и проявляется в виде личностных изменений.
Болезнь Кройцфельдта-Якоба
Прионовые белки млекопитающих не сходны с прионовыми белками дрожжей по аминокислотной последовательности. Несмотря на это, основные структурные особенности (формирование амилоидных волокон и высокая специфичность, препятствующая передаче прионов от одного вида организмов к другому) у них общие. Вместе с тем, прион, отвечающий за коровье бешенство, обладает способностью передаваться от вида к виду. В ходе исследований мозговых тканей умерших от прионных инфекций животных было показано, что прионы не содержат нуклеиновых кислот, а представляют собой белки. Одним из первых охарактеризованных прионных белков стал PrP (от англ. prion-related protein или protease-resistant protein) массой около 35 кДа. Известно, что PrP может существовать в двух конформациях — «здоровой» — PrPC, которую он имеет в нормальных клетках (C — от англ. cellular — «клеточный»), в которой преобладают альфа-спирали, и «патологической» — PrPSc, собственно прионной (Sc- от scrapie), для которой характерно наличие большого количества бета-тяжей. При попадании в здоровую клетку, PrPSc катализирует переход клеточного PrPC в прионную конформацию. Накопление прионного белка сопровождается его агрегацией, образованием высокоупорядоченных фибрил (амилоидов), что в конце концов приводит к гибели клетки. Высвободившийся прион, по-видимому, оказывается способен проникать в соседние клетки, также вызывая их гибель. Функции белка PrPC в здоровой клетке пока не определены. В норме белок PrPC ассоциирован с клеточной мембраной, гликозилирован остатком сиаловой кислоты. Он совершает циклические переходы внутрь клетки и обратно на поверхность в ходе эндо- и экзоцитоза. Один такой цикл длится около часа. В эндоцитозном пузырьке или на поверхности клетки молекула PrPC может разрезаться протеазами на две примерно равные части. До конца механизм спонтанного возникновения прионных инфекций не ясен. Считается (но ещё не полностью доказано), что прионы образуются в результате ошибок в биосинтезе белков. Мутации генов, кодирующих прионный белок (PrP), ошибки трансляции, процессы протеолиза — считаются главными кандидатами на механизм возникновения прионов. Согласно недавно проведённым исследованиям прионы способны к дарвиновской эволюции за счёт действия естественного отбора. Есть данные, дающее основание считать, что прионы являются не только инфекционными агентами, но и имеют функции в нормальных биопроцессах. Так, например, существует гипотеза, что через прионы осуществляется механизм генетически обусловленного стохастического старения. Человек может заразиться прионами, содержащимися в пище, так как они не разрушаются ферментами пищеварительного тракта. Беспрепятственно проникая через стенку тонкого кишечника, они в конечном итоге попадают в центральную нервную систему. Так переносится новый вариант болезни Крейтцфельдта-Якоба (nvCJD), которой люди заражаются после употребления в пищу говядины, содержащей нервную ткань из голов скота, больных бычьей губчатой энцефалопатией (BSE, коровье бешенство). Прионы могут проникать в тело и парентеральным путем. Были описаны случаи заражения при внутримышечном введении препаратов, изготовленных из человеческих гипофизов (главным образом гормоны роста для лечения карликовости), а также заражение мозга инструментами при нейрохирургических операциях, поскольку прионы устойчивы к применяемым в настоящее время термическим и химическим методам стерилизации. Эта форма болезни Крейтцфельдта-Якоба обозначается как ятрогенная (1CJD). При определённых, неизвестных условиях, в организме человека может произойти спонтанная трансформация прионного протеина в прион. Так возникает так называемая спорадическая болезнь Крейтцфельдта-Якоба (sCJD), впервые описанная в 1920 г. независимо друг от друга Гансом Герхардом Крейтцфельдтом и Альфонсом Марией Якобом. Предполагается, что спонтанное возникновение этой болезни связано с фактом, что в норме в человеческом теле постоянно возникает небольшое количество прионов, которые эффективно ликвидируются клеточным Аппаратом Гольджи. Нарушение этой способности «самоочищения» клеток может привести к повышению уровня прионов выше допустимой границы нормы и к их дальнейшему неконтролируемому распространению. Причиной возникновения спорадической болезни Крейтцфельдта-Якоба согласно этой теории является нарушение функции Аппарата Гольджи в клетках. Особую группу прионовых заболеваний представляют собой наследственные (врожденные) болезни, вызванные мутацией гена прионового протеина, который делает возникший прионовый протеин более подверженным спонтанному изменению пространственной конфигурации и превращения их в прионы. К этой группе наследственных заболеваний относится и наследственная форма болезни Крейтцфельдта-Якоба (fCJD), которая наблюдается в ряде стран мира. При прионовой патологии наивысшая концентрация прионов обнаружена в нервной ткани заражённых людей. Значительное количество прионов встречается в лимфатической ткани. Наличие прионов в биологических жидкостях, включая слюну, пока не было однозначно подтверждено. Если представление о постоянном возникновении небольшого количества прионов верно, то можно предположить, что новые, более чувствительные методы диагностики откроют это количество прионов, разбросанное по различным тканям. В данном случае, однако, речь пойдёт о «физиологическом» уровне прионов, которые не представляют собой никакой угрозы для человека.
Болезнь Крейтцфельдта-Якоба: симптомы
Болезнь Крейтцфельдта-Якоба имеет характерную симптоматику, независимо о происхождения. Ятрогенная, наследственная и классическая форма проявляется примерно одинаково. Несколько иначе протекает новый вид заболевания.
Основные проявления
Начальная стадия проявляется временными выпадениями из памяти событий, изменением настроения, потерей интереса к окружающим. Постепенно пациент испытывает все больше трудностей в осуществлении жизненно необходимых действий. Конечная стадия сопровождается нарушением зрения, медленной речью и появлением галлюцинаций.
Примерно у 40% больных со спорадической формой болезнь протекает в подостро, с постепенно усугубляющимися когнитивными отклонениями. Еще 40% зараженных страдают нарушениями функций мозжечка, а у остальных описывается смешанная форма заболевания.
Наиболее характерными клиническими симптомами классической болезни Крейтцфельдта-Якоба являются:
- нарушение поведения;
- ухудшение памяти;
- диплопия и развитие корковой слепоты;
- психические расстройства;
- дисфункция мозжечка, атаксия;
- нарушение координации;
- парезы;
- дизартрия;
- неспособность к самообслуживанию, неряшливость и другие признаки слабоумия;
- сочетание признаков поражения пирамидной и экстрапирамидной системы.
У многих пациентов отмечаются локальные или генерализованные виды эпилептических припадков. Они проявляются в ответ на определенные раздражители – свет, звук, прикосновение.
Терминальная стадия сопровождается глобальными когнитивными отклонениями, полной неспособностью к передвижению. Смерть обычно наступает в течение от 8 месяцев до 2 лет после появления начальных отчетливых симптомов заболевания. Новая форма болезни характеризуется выходящими на первый план психическими нарушениями и изменениями чувствительности.
Стадии
Специалисты выделяют три стадии протекания болезни Крейтцфельдта-Якоба:
- Продромальная стадия. Симптомы ее не имеют специфических проявлений и наблюдаются примерно у 30% всех заболевших. У человека отмечается астения, нарушение сна и аппетита, снижение способности к запоминанию, похудение, легкие когнитивные отклонения. Для пациента характерно появление фобий, бреда, галлююцинаций.
- Инициальная стадия. Начинаются изменения со стороны зрения, появляется головная боль, нарастает неустойчивость, атаксия, нарушение чувствительности, тремор, миоклонии, эпилептические припадки, и другие неврологические симптомы на фоне проявления слабоумия.
- Развернутая стадия сопровождается спастическим параличом, всеми признаками нарушений работы мозговых структур, тяжелой формой деменции без возможности контакта с больным. У человека полностью утрачивается контроль над работой тазовых органов, нарушается глотание, отмечается снижение температуры.
Чаще всего болезнь Крейтцфельдта-Якоба развивается постепенно, но иногда она имеет острое и подострое начало. Смерть наступает в результате отказа от работы жизненно важных центров и выраженного истощения.
Диагностика болезни Крейтцфельдта-Якоба
Предположительный диагноз болезни Крейтцфельдта-Якоба ставится на основании клинических проявлений и данный дополнительных методов исследования. Заподозрить патологию можно при сочетании определенных симптомов, наблюдающихся в течение продолжительного времени:
- прогрессирование деменции;
- пирамидальные и экстрапирамидальные нарушения;
- миоклонии;
- расстройства работы мозжечковых структур;
- нарушение зрения.
Для уточнения патологии невролог направляет человека на прохождение энцефалографии, МРТ мозга, спинномозговую пункцию, стереотаксическую биописию:
- На ЭЭГ у почти у каждого пациента с болезнью Крейтцфельдта-Якоба отмечается снижение электрической активности с периодически выскакивающими острыми волнами. Наблюдаются изменения, характерные для миоклонии, у 50% больных. Новый вид заболевания часто не проявляется какими-либо нарушениями на энцефалограмме.
- На МРТ можно выявить признаки атрофических изменений в мозжечке и коре мозга (синдром «медовых сот»), расширение желудочков. От подкорковых структур и таламуса иногда улавливаются участки с повышенным сигналом.
- Люмбальная пункция проводится в обязательном порядке при подозрении на болезнь Крейтцфельдта-Якоба. Исследование ликвора позволяет провести дифференциальный диагноз с другими патологическими нарушениями ЦНС.
- Самым объективным способом, который помогает достоверно определить диагноз, является морфологическое исследования биоптата ткани мозга. С помощью иммунохроматографического метода есть возможность обнаружить белок прион. Его наличие позволяет утверждать о том, что у больного наблюдается именно болезнь Крейтцфельдта-Якоба.
Проводить дифференциальную диагностику приходится с другими заболеваниями центральной нервной системы, которые протекают с похожими симптомами:
- старческая, мультиинфарктная деменция;
- болезнь Альцгеймера;
- энцефалопатия Хашимото;
- воспалительное поражение мозговых оболочек (энцефалит, арахноидит, менингит).
Диагностика
Диагностика болезни Крейтцфельдта-Якоба основывается на данных клинической картины, неврологического статуса, случаев употребления мяса животных из очагов эпидемий спонгиозной энцефалопатии животных.
Также необходимо исключение другой патологии, которая могла бы привести к клиническим проявлениям заболевания. Так, при ЭЭГ исследовании выявляются фоновые плоские колебания в виде волн, состоящих из трех фаз. На эти волны могут накладываться острые и медленные высокоамплитудные волны с частотой около 2 Гц.
МСКТ и МРТ исследование выявляет признаки дистрофии ткани мозга. Кровь и ликвор, как правило, не меняются. Достоверным диагноз считается после проведения прижизненной биопсии мозга с использованием специальных молекулярных методов исследования. В случае невозможности проведения исследования, несогласии больного или его законных представителей, диагноз считается вероятным, но не подтвержденным.
Лечение болезни Крейтцфельдта-Якоба в Москве
Этиотропная терапия заболевания до сих пор находится в разработке. Поэтому все лечение его в клинике направлено на купирование симптоматических проявлений. Пациенту отменяются все ранее применяемые препараты, поскольку они могут негативно отражаться на течении болезни, усугубляя нарушение мнестических функций и ухудшая поведение больного.
Консервативная терапия
Уже доказано, что традиционные средства, которые успешно используются при лечении вирусной инфекции (вакцины, интерфероны) не оказывают положительного действия на течение болезни Крейтцфельдта-Якоба. Некоторые результаты с временным улучшением состояния удается получить при введении антибиотика, который блокирует работу аппарата Гольджи внутри клетки и препятствует размножению прионов. Замечено, что при использовании некоторых блокаторов каналов кальция длительность функционирования пораженных структур мозга увеличивается по времени.
Некоторое замедление накопления патологических белков отмечаются при использовании иммуномодулятора, который стимулирует выработку эндогенного интерферона. Но эти препараты обладают сильными побочными действиями.
Симптоматическая терапия у больных направлена на устранение:
- тремора и приступов эпилепсии;
- экстрапирамидальных нарушений;
- депрессии и беспокойства;
- болевого синдрома.
Для этого используются различные группы препаратов, их выбор осуществляет врач. Основанием к применению того или иного средства является наличие и степень выраженности отклонения. В терапии применяются такие группы препаратов:
- нейролептики;
- транквилизаторы;
- анальгетики;
- опиаты;
- седативные средства;
- антидепрессанты;
- противосудорожные лекарства.
Преимущество нахождения больного в клинике
После установления диагноза болезни Крейтцфельдта-Якоба, человеку требуется обеспечить полноценную симптоматическую терапию, психологическую поддержку пациента и членов его семьи. Чем дальше прогрессирует патология, тем больше необходимо пациенту заботы и внимания, так как у него постепенно утрачиваются навыки самообслуживания, нарушается работа органов малого таза.
Работающие родственники не могут обеспечить круглосуточное дежурство возле пациента. А его потребуется кормить, мыть и помогает в передвижении по мере необходимости. В некоторых случаях ему приходится выводить мочу при помощи катетеризации в результате нарушения работы мочевого пузыря. А эту манипуляцию в состоянии выполнить только специалисты с медицинским образованием.
При нарушении функции глотания человеку потребуется питания при помощи специальной трубки и постановка капельных растворов с необходимыми веществами. В домашних условиях обеспечить полноценное лечение и уход возможно только на самых ранних стадиях. В последующем больному обязательно потребуется систематический уход специально подготовленного персонала.
В терминальной стадии проводятся мероприятия направленные на продление жизни. Их осуществляют только при наличии реанимационного оборудования – ИВЛ, оксигенотерапия, средства для стимуляции работы дыхательной и сердечно-сосудистой системы.
При выборе клиники для оказания помощи пациентам с болезнью Крейтцфельдта-Якоба, родственникам следует понимать, что наиболее качественная помощь может быть оказана в заведении, в котором работают опытные специалисты с определенным профилем.
Если госпитализировать человека в государственный стационар, то можно сэкономить средства, но при этом условия пребывания будут не идеальными:
- в палатах размещаются по нескольку человек;
- препараты придет приобретать самостоятельно, так как те, что есть в наличии не всегда могут эффективно устранять осложнения;
- у пациента есть высокий риск подхватить внутрибольничную инфекцию;
- персонал не сможет постоянно контролировать пациента и оказывать ему помощь при необходимости 24 часа в сутки.
Когда больной находится еще в сознании и способен оценивать сложившуюся ситуацию, его пребывание в госучреждении значительно усугубит состояние, вызовет депрессию и усиление психических нарушений.
КЛИНИЧЕСКОЕ НАБЛЮДЕНИЕ БОЛЕЗНИ КРЕЙТЦФЕЛЬДТА-ЯКОБА Толстолуцкая А.О.,Делинская Д.А.
Текст научной статьи
Введение: Прионные заболевания имеют длительный инкубационный период, злокачественное течение и приводят к быстрой гибели. Данные страдания вызывает аномальная разновидность прионов. Возбудителем прионных болезней является инфекционный прионный белок Prion Protein — scarpie (PrPSc), образующийся в результате конформационных изменений нормального (неинфекционного) клеточного белка Prion Protein — cellular (PrPс) [1, 58]. Накопление патологической формы PrPSc ведет к массовой гибели нервных клеток. Прионы — единственные известные инфекционные агенты, размножение которых происходит без участия нуклеиновых кислот. Вопрос о том, считать ли прионы формой жизни, в настоящий момент является открытым. Болезнь Крейтцфельдта-Якоба (БКЯ) относится к одной из четырех известных редких человеческих прионных болезней. Данное заболевание было описано в 1920-х годах Г. Г. Крейцфельдтом и А. М. Якобом. БКЯ — по праву одно из самых опасных инфекционных заболеваний, даже несмотря на свою крайне редкую распространенность: 0,5-1,5 случая на 1 млн населения. 90% всех случаев БКЯ приходится на классическую форму, чаще болеют мужчины [2, 68]. Общее количество зарегистрированных случаев варианта БКЯ в 7-ми странах Европы и 4-х других странах составило 219 наблюдений [3, 599]. Мы унифицировали существующие классификации в единую с разделением на эндогенные (спорадическая, наследственная и новая) и экзогенные (ятрогенная) формы. Ведущими симптомами в процессе наблюдения у больных были: нарастающие интеллектуально-мнестические расстройства, амнезия, страх, депрессия и деменция, дизартрия и дисфония, атаксия, экстрапирамидная ригидность, тетрапарез, амиотрофии, офтальмопарез, судорожный синдром, миоклонии, дрожание головы и конечностей, бессонница, гипертермия [4]. Международным Консорциумом по БКЯ были предложены определенные критерии для диагностики БКЯ. В данных критериях учитываются: I. Клинические симптомы: • Деменция. • Мозжечковые или зрительные. • Пирамидные или экстрапирамидные. • Акинетический мутизм. II. Данные дополнительных исследований: • Типичный волновой комплекс (феномен PSWC) на ЭЭГ. • Определение белка 14-3-3 в ЦСЖ (у пациентов с длительностью заболевания менее 2 лет). • Повышение интенсивности сигнала от хвостатого ядра и скорлупы или, по меньшей мере, от 2 корковых регионов (темпорально-париетально-окципитальный) в режиме DWI или FLAIR. Согласно этой схеме для постановки вероятного диагноза БКЯ необходимо наличие двух критериев из пункта I и, по меньшей мере, одного критерия из пункта II [5, 64]. Наибольшую опасность в плане развития эпидемии представляет ятрогенная форма. В 1974 году был впервые зарегистрирован случай развития БКЯ у пациента через 18 месяцев после пересадки роговицы от донора с установленной БКЯ. Доказанные способы передачи: трансплантация донорской зараженной роговицы и твердой мозговой оболочки, лечение препаратами человеческого гормона роста и гонадотропинов, использование инфицированных инструментов и внутримозговых электродов при нейрохирургических операциях. Описаны единичные случаи БКЯ среди нейрохирургов, патологоанатомов и ветеринарных работников. Инкубационный период колебался от 4 до 30 лет [6, 2661]. Нами был описан случай БКЯ у пациента с пересадкой плечевой кости в 18летнем возрасте. Клинический случай: Больной Л. 31 год, поступил в клинику неврологии РостГМУ с грубыми психическими и поведенческими расстройствами. Жалоб не предъявлял. Со слов родственников, неврологические симптомы появились с июля 2021 года. При обращении к врачу состояние больного было расценено как цереброастенический синдром. Была назначена терапия без выраженного положительного эффекта. В августе 2021 года была проведена магнитно-резонансная томография (МРТ): МР картина единичных локальных арахноидальных изменений ликворокистозного характера, убедительных МР-данных за наличие изменений очагового характера в веществе мозга не выявлено. В сентябре 2021 года присоединились насильственные движения в руках, нарушения сна, речевые нарушения в виде эхолалии, больному стало трудно подбирать слова, он стал неопрятен, забывал о приеме пищи. Грубо нарушилась походка по типу апраксии ходьбы. Со слов родственников в течение сентября 2021 года потерял навыки самообслуживания, утратил речевой контакт, не мог выполнить задания по команде и по подражанию. Больной был проконсультирован психиатром, поставлен диагноз: диссоциативное расстройство по типу псевдодеменции. Из анамнеза жизни: пересадка плечевой кости в 1996 году. Был проведен ряд исследований: Электроэнцефалография (ЭЭГ): Уплощенный тип ЭЭГ. Значительные диффузные биоэлектрические активности головного мозга регуляторного характера. Снижение функциональной лабильности коры. Типичная эпилептиформная активность не зарегистрирована. МРТ головного мозга: МР-признаки умеренной дилатации субарахноидальных пространств. На магнитно-резонансной ангиографии (МРА) патологии сосудов артериального круга не выявлено. Биохимическое исследование крови: билирубин общий (39.2 мкмоль/л), мочевина (7.7 ммоль/л) повышены. Ультразвуковое исследование (УЗИ) почек: признаки умеренных диффузных изменений паренхимы и чашечно-лоханочной системы (ЧЛС). Церулоплазмин: 0.218 г/л (N = 0.2-0.6 г/л). Консультация офтальмолога: Ангиопатия сосудов сетчатки обоих глаз, данных за наличие колец Кайзера-Флешнера на роговице, отека сетчатки, застойных дисков зрительных нервов нет. УЗИ органов брюшной полости: незначительные диффузные изменения паренхимы поджелудочной железы. Нечеткие признаки холецистопанкреатита. Гастроэнтеролог: нельзя исключить гепатоцеребральную дегенерацию. Рекомендован биохимический анализ крови (аланинаминотрансфераза (АЛТ), аспартатаминотрансфераза (АСТ), щелочная фосфатаза, билирубин, мочевина) в динамике. Поступил в клинику неврологии для уточнения диагноза. Неврологический статус: не ориентирован в месте, времени, собственной личности. Не доступен продуктивному вербальному контакту. При проверке высших мозговых функций была выявлена грубая сенсорная афазия, моторная апраксия поэтому диагностика других корковых нарушений не представлялась возможной. Оценка функции фонации и артикуляции затруднена. Глоточные рефлексы проверить невозможно из-за активного сопротивления больного, язык — самостоятельно не показывает, при попытке высунуть — быстро прячет обратно. Вызываются рефлексы орального автоматизма (ладонно-подбородочный, хоботковый). Объем активных движений в суставах верхних конечностей снижен. Феномен паратонии. Корковые миоклонии. Мышечная сила в конечностях до 5 баллов. Хватательные автоматизмы. Пассивные движения: мышечный тонус повышен по экстрапирамидному типу, больше справа. Сухожильные рефлексы с верхних и нижних конечностей высокие D>S, с расширением рефлексогенных зон, без патологических стопных знаков. Оценить чувствительные нарушения не представляется возможным, в позе Ромберга — не устойчив с закрытыми и открытыми глазами. Пальцеколенную пробу и пальценосовую пробу не выполняет, так как не понимает задание. Нистагма нет. Тазовые функции контролирует. Менингеальные симптомы: отрицательны. Было проведено повторное ЭЭГ: уплощенная кривая. Реактивность мозговых систем резко снижена. При проведении провоцирующих ФП на ФН 3-27 замедление биоэлектрической активности в левой затылочно-теменной и правой передневисочной областях. При проведении фотостимуляционных проб на ФТ-2, ФТ-6, ФТ-10 отмечается замедление биоэлектрической активности по левому полушарию с акцентом в теменно-центрально-лобную область. Учитывая молодой возраст пациента, короткий анамнез, быстрое развитие симптомов в течение трех месяцев, наличие лобного синдрома (эйфоричность, некритичность к своему состоянию, дурашливость), поражения экстрапирамидной системы (повышение мышечного тонуса по пластическому типу), грубые когнитивные нарушения (псевдодеменция), анамнестические данные (пересадка плечевой кости в 1996 году), исключение СПИД-дементного комплекса и данные ЭЭГ (уплощение кривой и отсутствие эпилептических комплексов), был предположен диагноз: болезнь Крейтцфельдта-Якоба. Несмотря на выполненную МРТ головного мозга для подтверждения диагноза была повторно сделана МРТ в режиме DWI: МР-признаки поражения коры больших полушарий и подкорковых ядер головного мозга слева. Изменения характерные для губчатой энцефалопатии (болезнь Крейтцфельдта-Якоба). Выводы: • Отсутствие настороженности практикующих врачей в отношении редких фатальных опасных инфекционных заболеваний при наличии скоротечно развивающейся деменции в молодом и среднем возрасте. • Целесообразность выполнения МРТ в режимах DWI и FLAIR для подтверждения диагноза «БКЯ». • Возможность инфицирования больных от трупного костного материала. • Необходимость в разработке четкого плана лечения после постановки диагноза.
Вопросы и ответы
Если находится в постоянном контакте с человеком, у которого болезнь Крейтцфельдта-Якоба, есть ли шанс заразиться?
Заражение при обычном контакте происходит крайне редко. Но во время ухода следует придерживаться гигиенических правил. Они заключатся в мытье рук перед едой, защите кожи от попадания биологических жидкостей, закрытии всех раневых поверхностей водонепроницаемыми повязками.
Можно ли вылечить человека при этом заболевании?
Данное заболевание не поддается лечению и приводит к летальному исходу со 100% степенью вероятности. Все что можно предпринять в этом случае – обеспечить достойный уход за пациентом, купировать возникающие осложнения и проводить симптоматическую терапию с целью повышение качества жизни.
Почему болезнь Крейтцфельдта-Якоба приводит к смерти?
По мере прогрессирования патологии развиваются различные осложнения, которые чаще всего становятся причиной смерти. Некоторые из них поддаются медикаментозной коррекции – эпилептические приступы, острые психозы, пневмонии из-за присоединения инфекции. Правильный уход может снизить страдания человека от появления пролежней в результате дегенерации мышц. Финалом заболевания становится развитие дыхательной или сердечно-сосудистой недостаточности, которые заканчиваются летальным исходом. Но даже эти функции можно поддерживать еще некоторое время с помощью специальных приборов.