Epileptic encephalopathy is a progressive disorder of brain function against the background of clinically manifested or non-convulsive epilepsy. More often found in childhood. In adults, it manifests itself as affect disorders, personality changes, and sometimes addictions.
offers professional help for epilepsy. Call tel. 8(969)060-93-93.
Causes
It is based on organic changes in cerebral structures caused by genetic and acquired influences. Provoking factors in children are considered:
- Unfavorable heredity. Infantile epileptic encephalopathy is found in certain chromosomal abnormalities and gene mutations.
- Metabolism failures. EE is potentiated by propionic acidemia, methylmalonic aciduria, necrotizing hyperhycinemia and some other congenital metabolic pathologies.
- Brain tumors. The risk of developing the disease depends on the location and type of tumor.
- Developmental anomalies. The cause is focal cortical dysplasia, cerebral dysgenesis, Ecardi syndrome, porencephaly, hemimegalencephaly.
- CNS lesions. The disease is detected when the brain is damaged in prenatal, infancy and early childhood.
Other unfavorable circumstances in all categories of patients include pathologies of pregnancy, prematurity, mental disorders and chemical dependencies in parents, epileptoid syndromes in relatives, mild TBI and febrile convulsions.
MODERN ASPECTS OF GENETICS OF EPILEPTIC ENCEPHALOPATHY AND SYMPTOMATIC EPILEPSY IN CHILDREN
Epilepsy is one of the most pressing problems in pediatric neurology. The prevalence of epilepsy in the population is quite high and amounts to 0.5-0.75%, and among the child population it reaches 1%. Approximately 7% of children experience at least one seizure during their lifetime. [13].
According to Alimova V.S. (2005), which studied the frequency and prevalence of epilepsy among children, adolescents and adults of the Republic of Uzbekistan for the period 2001-2003, revealed that in 2003, patients with epilepsy accounted for 5.45% of all neurological patients. At the same time, the incidence rate of epilepsy ranged from 1.52 per 1000 population in 2001 to 1.12 in 2003. The author notes that during the period under study, the incidence among children and adolescents significantly exceeded these indicators among the adult population. [2,5].
From a genetic point of view, all forms of epilepsy can be divided into monogenic diseases and diseases with a hereditary predisposition. According to the latest achievements of molecular genetic studies of various forms of epilepsy, several groups of genes involved in the development of idiopathic epilepsy are identified [8, 10]:
- genes of potential-dependent ion channels, which include, first of all, the genes of sodium, potassium, calcium and chlorine channels.
Recent advances in molecular genetic research into epilepsy have revealed that some forms of epilepsy are based on disruption of the activity of ion channels. It is likely that their widespread distribution in the brain, as well as mutations in them, contribute to the occurrence of seizure activity. Disturbances in these genes can directly or indirectly lead to abnormal increases in neuronal excitability. [6, 7].
- genes for receptors and transporters of neurotransmitters of inhibition and excitation (gamma-aminobutyric acid, serotonin, glutamate).
Despite the fact that in recent years active molecular genetic studies of epilepsy have been carried out all over the world and more than a dozen genes are already known, mutations in which lead to the development of various forms of epilepsy, there is still a lot of uncertainty in this area, as well as in epileptogenesis in general. [14,16].
Thus, uncovering the genetic aspects of epilepsy is necessary for a clear understanding of the etiology and pathogenesis of this disease and the search for new ways of correction. However, the specific role of genetic factors in epilepsy remains unknown, especially among Uzbek children.
In general, in recent years, with the use of molecular technologies, obvious significant progress has been made in clarifying the molecular genetic basis of epilepsy. Molecular differentiation of disease cases provides clinicians with new diagnostic tools for reliable diagnosis, allowing them to accurately assess genetic risk, provide prognosis during counseling, and plan rational therapeutic approaches.
To identify mutations and polymorphisms in the studied patients with epileptic encephalopathy, we analyzed the gene a 1 subunit of the neuronal sodium channel (SCN1A), in which mutations and polymorphisms were previously identified in patients with various forms of epilepsy.
The SCN1A gene, encoding the al-subunit of the neuronal sodium channel, covers about 70,000 base pairs (bp) of genomic DNA, includes 26 exons (length from 8 bp to 3260 bp) and introns located between them [ 18]. The SCN1A gene product consists of four homologous domains containing six transmembrane segments and connected by intracellular loops. The unique sequence of the S4 segment carries a positive residue at every third position of the transmembrane segment. It is assumed that this property ensures the sensitivity of the channel to the electric field. The structure forming the ion pore is located between the highly conserved S5 and S6 segments [19].
The SCN1A gene belongs to a family of genes that provide instructions for sodium channels. These channels, which transport positively charged sodium atoms (sodium ions) into cells, play a key role in the cell's ability to generate and transmit electrical signals.
The SCN1A gene contains instructions for making one part (the alpha subunit) of sodium channels called NaV1.1. These channels are found in the brain and muscles, where they control the flow of sodium ions into the cell. In the brain, NaV1.1 channels are involved in transmitting signals from one nerve cell (neuron) to another. Communication between neurons depends on chemicals called neurotransmitters, which are released from one neuron and picked up by neighboring neurons. The flow of sodium ions through NaV1.1 channels helps determine when neurotransmitters will be released.
More than 150 mutations in the SCN1A gene have been associated with various grasping disorders that begin in infancy or childhood. Some of these conditions are relatively mild. These conditions include simple febrile (fever-related) seizures, which begin in infancy and usually stop by age 5, and generalized epilepsy with febrile seizures plus (GEFS+). GEFS+ includes febrile and other types of seizures that may persist beyond childhood. Other conditions cause more severe attacks that last longer and may be difficult to control. These repeated attacks can worsen over time and lead to decreased brain function. Severe seizure disorders caused by SCN1A mutations include severe myoclonic epilepsy of infancy (SMEI) and intractable epilepsy of childhood with generalized tonic-clonic seizures (ICE-GTC). [12].
SCN1A mutations underlying epileptic seizures have variable effects on NaV1.1 channel function. Mild disorders are caused by mutations that alter a single amino acid in the channel, which alter the structure of the channel. More severe uptake disorders may result from various changes in the SCN1A gene. Some mutations result in the production of non-functional versions of the NaV1.1 channel or reduce the number of these channels produced in each cell. Other mutations change single amino acids in critical regions of the channel. All of these genetic changes affect the ability of NaV1.1 channels to transport sodium ions in neurons. It is unclear, however, why these genetic changes lead to such a large range of seizures.
A common change (polymorphism) in the SCN1A gene has been associated with the effectiveness of certain anti-uptake drugs. This polymorphism, which is written as ICS5N +5 G>A, changes one DNA building block (nucleotide) in the SCN1A gene. Research suggests that this polymorphism is associated with the maximum safe amount (dose) of the anti-seizure drugs phenytoin and carbamazepine. These drugs treat epilepsy by blocking sodium channels (eg NaV1.1) in neurons. A dose that is too low may not effectively control seizures, and a dose that is too high may cause unwanted side effects. For now, doctors must use trial and error to determine the right dose for each person. The researchers hope that doctors will one day be able to use the ICS5N +5 G>A polymorphism to determine the safest and most effective doses of anti-uptake drugs for each person.
Cytogenetic location of the SCN1A gene - 2q24.3
Molecular Location on Chromosome 2: 166,845,669 base pairs to 167,005,641 (Figure 1)
Rice. 1. The SCN1A gene is located on the long (e) arm of chromosome 2 at position 24.3.
More precisely, the SCN1A gene is located from base pairs 166,845,669 to base pairs 167,005,641 on chromosome 2.
Currently, more than 70 mutations in the SCN1A gene are known, causing the development of such forms of epilepsy as benign familial seizures of newborns, generalized epilepsy with febrile seizures plus and febrile seizures [14].
Mutations leading to the development of benign familial seizures of early childhood are nonsense mutations, splice site mutations, frameshift mutations and are mainly found in the S5-S6 segments and the loop between them. Most often, they completely disrupt the function of the neuronal sodium channel [5].
The “milder” phenotype of generalized febrile seizures plus (GEFS+) is caused by missense mutations in the loop connecting the S2 and S3 segments of the SCN1A gene, leading to an increase in the time it takes to restore channel activity after inactivation [8,11]
Of the entire sample of patients with epileptic encephalopathy in children (n=20), polymorphism 3184 A–G was found in the homozygous state (3184*A/*A) in 8 patients (40%), in the heterozygous state (3184*A/*G) - in 11 (56%), and the homozygous genotype (3184*G/*G) was determined only in 1 patient (4%) (Fig. 2).
Rice. 2. Frequency of occurrence of polymorphism 3184 A– G in children with epileptic encephalopathy
Thus, we found that a number of linkage loci were mapped for epileptic encephalopathy: 40% - 168bp; 56% - 168, 145 and 23bp and 4% -145 and 23 bp (Fig. 3).
Rice. 3. Mapped series of linkage loci in children with epileptic encephalopathy of Uzbek nationality
As can be seen from the data obtained, in the majority of patients with epileptic encephalopathy, mutations were found in the SCN1A gene, encoding the α1 subunit of the neuronal sodium channel, which causes an increase in the recovery time of the channel activity after inactivation, which in turn also leads to hyperexcitability of the neuron.
In the process of development of this form of the disease, the participation of several interacting genes located in these areas is assumed. The contribution of each of them to the risk of developing epileptic encephalopathy may be small, but, nevertheless, leads to heritable changes that predispose to seizure activity.
Thus, taking into account all of the above, we can draw the following conclusion: mutations and polymorphisms in the SCN1A gene of the neuronal sodium channel lead to an increase in the recovery time of channel activity after inactivation and, as a result, provoke hyperexcitability of neurons. The changes in the nucleotide sequence of the SCN1A gene that we discovered may make a certain contribution to the development of predisposition to epileptic encephalopathy in children of Uzbek nationality.
- Akre B., Rasmussen M; Lundby R. Pyridoxine – dependent seizures //Norwegian Journal In formation. -2004. -R. 162-164.
- Alkan A., Kutlu R., Aslan M. Pyridoxine – Dependent Seizures: Magnetic Resonance Spectroscopy Findings // Journal of Child Neurology. -2004. Vol. 19, no. 1. -R
- Battaglioli G., Rosen DR, Gospe SM Jr., Glutamate decarboxylase is not denetically linked to pyridoxine-dependent seizures // Neurology. -2000. -No. 25. 55(2). -309-311.
- Berkovic S., Howell R., Hay D. Epilepsies in twins: genetics of the major epilepsy syndromes II Neurol. - 1998. - V. 43. - P. 435-445.
- Biervert C, Schroeder B, Kubisch C, et al. A potassium channel mutation in neonatal human epilepsy II Science - 1998. - 279. - P. 403-409.
- Caraballo R., Garro F., Cersosimo R. Pyridoxine dependence: the importance of clinical diagnosis and early treatment // Revista de neurologia—2004. -No. 38. -P. 49-52.
- Jang YM, Kim DW, Kang TC Human pyridoxal phosphatase. Molecular cloning, functional expression, and tissue distribution // Journal of biological chemistry. -2003. -No. 12. -R. 5040-5046.
- Kaelin A., Caser JP, Jaeger R. Vitamin B6 metabolites in idiopathic calcium stone formers: no evidence for a link to hyperoxaluria // Urological research. -2004. -No. 32 (1). -P. 61-68.
- Kannan K., Jain SK Effect of vitamin B6 on oxygen radicals, mitochondrial membrane potential, and lipid peroxidation in H2 O2 treated U 937 monocites // Free radical biology medicine. -2004. -No. 15;36 (4). -P. 423-428.
- Kholin AA, Lemeshko ID, Il'ina ES, Voronkova KV, Petrukhin AS Seizure characteristics of early myoclonic encephalopathy // Extended Abstracts. 2nd East Mediterranean Epilepsy Congress. Dubai.-2010.- A-246-0004-00036.
- Kholin A.A., Voronkova K., Aivazjan S., Shevchenko A., Lemeshko I., Il'ina ES, Petrukhin AS Features of status epilepticus at infancy and early childhood // 28th International Epilepsy Congress 28th June-2nd July 2009.- P .757.
- Maeda T., Inutsuka M., Goto K., Irumi T. Transient nonketotic hyperglycinemia in an asphyxiated patient with pyridoxine-dependent seizures // Pediatr. Neurol. — -No. 22 (2). -P. 225-227.
- Najafi Reza Mohammad, Tamizifar Babak. Drug discontinuation in epileptic children: predictive value of the EEG // Archives of Iranian Medicine. -2002. -Vol. 5, No. 3, - P. 157-161.
- Okuda T., Sumiya T., Iwai N., Viyata T. Pyridoxine-5-phosphate oxidase is a candidate gene responsible for hypertension in Dohl's rats //Biochemical and biophysical research communications. -2004. -No. 313 (3). -P. 647-653.
- Said HM Recent advances in carrier-mediated intestinal absorption of water-soluble vitamins //Annual review of physiology. -2004. -No. 66. -P. 419-446.
- Schulre-Bonhage A., Kurthen M., Walger P. Pharmacorefractory status epilepticus due to low vitamin B6 levels during pregnancy //J. Epilepsy. -2004. -No. 45(1). -P.81-84.
- Tsuge H., Hotta N., Hayakawa T. Effects of vitamin B6 on (n-3) polyunsaturated fatty acid metabolism //J. Nutr. -2000. -No. 130. -P. 333-334.
- Voronkova K., Kholin A.A., Lemeshko I., Il'ina ES, Petrukhin AS Seizure characteristics of Dravet Syndrome // 28th International Epilepsy Congress 28th June-2nd July 2009.- P.750.
- Wanden LR Magnesium absorption using stable isotope tracers in healthy children and children treated for leukemia //Nutrition. -2001. -No. 3. -P. 221-224.
Classification
Taking into account the time of occurrence, the characteristics of the course and the prevailing symptoms, two types of EE are distinguished:
- First. Early epileptic encephalopathy. The course is severe, worsening impairments of speech, intelligence, and cognitive abilities prevail. The group includes Lennox and Ohtahara syndromes, infantile spasm, etc.
- Second. More often diagnosed in adults. There are no paroxysms, epi-activity is detected only according to additional studies. Behavioral, cognitive, mental and social impairments are observed.
Etiological factors
The causes of early epileptic encephalopathy are varied. They can be divided into genetic and non-genetic. The first group includes:
- developmental defects and perinatal diseases that cause structural damage to the brain;
- diseases that are accompanied by metabolic disorders (mitochondrial diseases, deficiency of the enzyme sulfate oxidase, phenylketonuria, etc.);
- other conditions in which damage to brain structures occurs (stroke, cortical dysplasia, trauma, tuberous sclerosis, infectious diseases).
The genetic causes of epileptic encephalopathy include mutations in genes, of which there are more than 35: STXBP1, KCNQ2, CASK, SCN2A, SLC25A22, STK9, PLCB1, etc. The disease can also be combined with chromosomal abnormalities such as Down syndrome or Angelman syndrome. According to the type of inheritance, epileptic encephalopathies can be X-linked dominant and recessive, as well as autosomal dominant and autosomal recessive.
Symptoms in children
The list of complications of the first kind includes changes in the psyche and behavior, neuropsychological disorders characteristic of organic diseases of the central nervous system.
Infants experience frequent regurgitation, problems with breastfeeding, poor sleep, increased muscle tone, and interruptions in heart function. Inappropriate reactions to external stimuli (sounds, light, changes in body position), anxiety, whims, and frequent crying are possible. Preschoolers continue to experience increased muscle tone, sleep problems, fainting and severe headaches. In the emotional sphere, frequent attacks of anger, mood instability, and uncertainty are noted; in the cognitive sphere, difficulties in switching attention and poor memorization are noted. The formation of epileptoid rigidity begins.
In school-age children, cognitive disorders come to the fore. Patients remember the material poorly, cannot cope with tasks, and have difficulty adapting to new rules and surroundings. Dizziness, headaches, and fainting persist. The formation of the epileptoid personality type is completed.
EE causes complex disorders of the psyche, speech, physical development and motor skills. With a progressive course, hydrocephalus, mental retardation, dementia, depression, mania, and psychosis are possible. The inability to learn results in social maladaptation, the inability to obtain a profession, and the need for outside care. With timely initiation of treatment, EE is sufficiently compensated for, children graduate from school, work, and start families.
Diagnostics
Performed by a neurologist, it involves a comprehensive examination using the following methods:
- Neurological examination. Includes the study of reflexes, muscle strength, various types of sensitivity.
- Neuroimaging. Electroencephalograms show areas of epiactivity. MRI may show signs of structural changes. Based on duplex scanning and Doppler sonography, cerebral circulation is assessed.
- Consultation with a psychiatrist. Involves the study of characterological, cognitive, behavioral characteristics, clarification of the severity of mental disorders.
- Psychological tests. Include questionnaires, projective methods, pathopsychological examination.
- Lab tests. Informative for some hereditary and metabolic variants of the disorder.
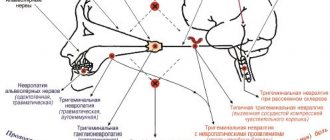
The nature of mental and neurological manifestations correlates with the location of the epifocus on the EEG. When localized in the left hemisphere, speech, writing and counting disorders predominate; in the right - disarticulation, inability to regulate and recognize changes in tone, rhythm and intonation in a conversation, difficulties in determining the location and size of objects. When the median structures are damaged, eccentric, antisocial or aggressive behavior and autism are noted.
In the absence of seizures, it can be difficult to determine that it is in adults. The epileptic nature of encephalopathy is supported by the coincidence of the location of the lesion with disorders of certain mental functions, the connection between epiactivity and the appearance of symptoms, and improvement in the condition when taking antiepileptic drugs.
Cost of services
| CONSULTATIONS OF SPECIALISTS | |
| Initial consultation with a psychiatrist (60 min.) | 6,000 rub. |
| Repeated consultation | 5,000 rub. |
| Consultation with a psychiatrist-narcologist (60 min.) | 5,000 rub. |
| Consultation with a psychologist | 3,500 rub. |
| Consultation with Gromova E.V. (50 minutes) | 12,000 rub. |
| PSYCHOTHERAPY | |
| Psychotherapy (session) | 7,000 rub. |
| Psychotherapy (5 sessions) | 30,000 rub. |
| Psychotherapy (10 sessions) | 60,000 rub. |
| Group psychotherapy (3-7 people) | 3,500 rub. |
| Psychotherapy session with E.V. Gromova (50 minutes) | 12,000 rub. |
| TREATMENT IN A HOSPITAL | |
| Ward for 4 persons | 10,000 rub./day |
| Ward for 3 persons | 13,000 rub./day |
| Ward 1 bed VIP | 23,000 rub./day |
| Individual post | 5,000 rub. |
| PETE | 15,000 rub./day |
This list does not contain all prices for services provided by our clinic. The full price list can be found on the “Prices” , or by calling: 8(969)060-93-93. Initial consultation is FREE!
Treatment
The treatment program for epileptic encephalopathies is compiled individually, taking into account age, type and causes of the disease. The following medications are used as part of EE therapy:
- Anticonvulsants. Relaxes muscles, eliminates convulsive attacks. Valproic acid is usually used as first-line drugs.
- Tranquilizers. Indicated for patients with minor seizures.
- Sedatives. Help stabilize the emotional state.
- Vasodilators. Improve blood flow in cerebral structures.
- Nootropics. Increases the activity of brain cells, promotes the development of memory and attention.
- Vitamins, amino acids. Stimulates metabolic processes in nervous tissue.
Additionally, non-drug methods are used. Children are offered psychocorrectional classes and work with a speech therapist. All patients benefit from exercise therapy and massage. It is recommended to normalize the daily routine, thoughtful alternation of loads and rest periods. The prognosis is determined by the form of the disease, the timeliness and adequacy of medical care.
You can make an appointment with a neurologist at the Leto clinic by phone. 8(969)060-93-93.
Modern principles of treatment of epileptic encephalopathy in children
The article presents a literature review of modern principles of treatment of epileptic encephalopathy. The main indicator of the correct choice of drug, dose and treatment success is the normalization of neurophysiological data. Clinical improvement in most cases is manifested by a certain, often significant lag relative to electroencephalography data. As has already been reported, another opportunity to improve the effectiveness and tolerability of antiepileptic therapy is the creation of new dosage forms of drugs that already exist in certain dosage forms, most often tablets.
The article presents a review of the literature of modern principles of treatment of epileptic encephalopathy. The main indicator of the correctness of the choice of drug, dosage and treatment success is normalization of neurophysiological data. Clinical improvement in most cases manifested a certain, often significant delay relative to the data of electroencephalography. As previously reported, another opportunity for improvements in the efficacy and tolerability of antiepileptic therapy — development of new dosage forms of drugs that already exist in certain dosage forms, more often — in the tablets.
The search for opportunities to increase the effectiveness of antiepileptic drugs (AEDs) includes: the synthesis of new drug formulas or modification of already synthesized molecules, the identification of new indications for use, the search for new dosage forms, new rational combinations of drugs, and others. Modern methods for solving the problem of side effects of AEDs include: prevention (prescribing AEDs taking into account the general somatic and mental health of the patient and his immediate family), correction of side effects, monitoring of the condition: timely examinations, consultations with specialists [7. 29].
The positive effect in most patients of antiepileptic therapy aimed at suppressing epileptic activity on the EEG, as well as the close temporal relationship between the regression of such epileptic activity and clinical symptoms confirm the key role of epileptic brain activity in the development of stable psychiatric and behavioral symptoms [4].
The failure of the use of neuroleptics and antidepressants for psychoses associated with epileptic discharges in the limbic-reticular system, as noted in the literature [6], as well as the effectiveness of antiepileptic drugs for them, are determined in 80% of patients, which corresponds to our data. By lowering the threshold of convulsive readiness and, accordingly, activating epileptic activity, antidepressants and antipsychotics not only have no effect, but even, as was the case in our patients, aggravate symptoms [12].
It should be noted that mental disorders (anxiety, aggressiveness, disorientation, memory and thinking disorders) regress much faster, usually on the 3rd–5th day with a stable therapeutic dose of the antiepileptic drug, which is apparently determined by the normalization of the limbic neuronal systems -reticular complex after the cessation of epileptic activity. Restoration of speech, cognitive processes, and social adaptation occur gradually over weeks, months and years, since these skills require learning in the course of everyday life [14].
The drug of first choice in the treatment of epileptic non-paroxysmal disorders and encephalopathies is valproic acid (Depakine Chrono, Depakine Chronosphere) [2, 10, 18, 23]. It suppresses epileptic activity in the brain. Normalization of the EEG begins already at subtherapeutic doses, which makes it possible on the 3rd–5th day of selecting a therapeutic dose to verify the correctness of the choice of drug, and subsequently, based on the dynamics of the EEG, adjust the dose and monitor the success of treatment [15]. Normalization in the EEG correlates with clinical improvement, which is especially important since patients do not have seizures, the dynamics of which could be used to monitor their effectiveness, and clinical normalization of mental and behavioral symptoms usually lags behind EEG normalization [3. 21]. In addition to its antiepileptic effect, valproic acid has its own antipsychotic effect.
In the USA, valproic acid accounts for 35% of all drugs prescribed for schizophrenia [22]. The main mechanism of action of valproic acid is an increase in GABAergic inhibition. By suppressing excessive discharge activity in the brain, it prevents excitotoxic neuronal death, which is one of the mechanisms of its neuroprotective action. The normalizing effect on kinases, gene expression and transcription leads to the normalization of plastic neuronal processes, which contributes to the interruption of the epileptic process and the patient’s recovery [20].
When applied to the treatment of childhood and infant epileptic encephalopathies, which are based on brain immaturity, it is important that valproic acid activates the growth of dendrites and axons in neuroblasts and the formation of a well-organized neural network from them, thereby facilitating the maturation of functional systems [13]. These data are in good agreement with the accelerated maturation of EEG parameters observed during the treatment of non-paroxysmal epileptic disorders with valproic acid [27]. The condition for effective treatment of epileptic encephalopathies is the stability of suppression of epileptiform activity, achieved by consistently maintaining the effective concentration of the drug in plasma.
Fejerman N. et al. [23], based on a synthesis of material from four clinics, came to the conclusion that levetiracetam is the most effective first-choice drug for this form. Accordingly, levetiracetam has been shown to be effective in the treatment of epileptic disorders of speech, behavior, memory and psyche, and general developmental disorders in patients without seizures [31]. The experiment demonstrated the ability of levetiracetam to counteract kindling in the amygdala, which determines its potential effectiveness in preventing the progression of epileptic encephalopathy [16].
Lamotrigine (Lamictal) is recommended for the treatment of infant and childhood catastrophic epileptic syndromes: West, Lennox-Gastaut, with persistent spike-wave complexes in slow-wave sleep and other non-paroxysmal epileptic disorders. Its use in non-paroxysmal epileptic disorders and epileptic encephalopathies is determined by the fact that it suppresses epileptiform activity in the EEG [17]. Limitations of lamotrigine as a first-line drug for the treatment of epileptic encephalopathies are due to its slow titration so that therapeutic concentrations are achieved weeks after initiation of treatment, and the fact that it may in some cases worsen seizures, epileptiform EEG activity, and cognitive and behavioral impairment. [1].
The effectiveness of topiramate has been shown in catastrophic epileptic encephalopathies - Lennox-Gastaut and West syndrome. Data on the successful treatment of epilepsy with electrical status epilepticus in slow-wave sleep indicate the possibility of its use in cognitive and mental disorders caused by this condition. There are also isolated reports of its use in epileptic disorders with autistic regression and learning disabilities [9]. However, the main side effect of topiramate is its negative impact on mental, behavioral and cognitive functions in some patients, which requires caution regarding the possibility of its use in mental and behavioral epileptic disorders, especially in patients with impaired verbal functions, which it itself can cause [10].
Benzodiazepines (clonazepam, clobazam, diazepam) and ethosuximide (Suxilep) are used as additional therapy for epileptic encephalopathies in cases resistant to monotherapy [12]. In severe forms of epileptic encephalopathies and non-paroxysmal mental, behavioral and neuropsychological epileptic disorders (West, Lennox-Gastaut, Landau-Kleffner syndromes, epilepsy with constant spike-wave complexes in sleep, opercular status epilepticus, epileptic speech dyspraxia), hormonal therapy is used, most often adrenocorticotropic hormone (ACTH) drugs. Both natural ACTH and its synthetic analogue Synacthen Depot, as well as prednisolone, are used. Drug dosages vary widely as individual patients respond to different treatment regimens [8, 19].
New AED formulas with new mechanisms of action are widely represented on the pharmaceutical market.
In Russia in 2010, lacosamide (Vimpat) was registered for the treatment of focal epilepsies in patients over 16 years of age in combination therapy. Lacosamide has a new antiepileptic mechanism. The drug selectively enhances slow inactivation of sodium channels without affecting fast inactivation, resulting in: stabilization of hyperexcitable neuronal membranes, normalization of activation thresholds and inhibition of repetitive neuronal discharges. Currently, both a tablet form of lacosamide and an intravenous solution are available, which is an alternative for patients temporarily unable to take the drug orally [11].
Other newer antiepileptic drugs are currently under registration: zonisamide (Zonegran) capsules as an additional drug in adults for the treatment of partial epileptic seizures with and without secondary generalization and rufinamide (inovelon) tablets as an additional drug in the treatment of epileptic seizures associated with Lennox-Gastaut syndrome in patients over 4 years of age. Zonisamide's mechanism of action is complex, but not fully disclosed. It is thought to act on voltage-gated sodium and calcium channels and also reduce neuronal activity via gamma-aminobutyric acid.
Rufinamide is a natium channel modulator. It was registered in the USA in 2004, in Europe - 2007. Clinical studies have convincingly proven its effectiveness in Dropatak therapy (reduces them by 42.5%, and the effect becomes noticeable in the first 2 weeks). Recently, it was shown that rufinamide can be effective in the complex treatment of epileptic encephalopathies and epileptic syndromes, in addition to Lennox-Gastaut syndrome, Dravet syndrome, malignant migratory partial seizures of infancy and others [24].
New indications for the use of certain AEDs have been approved. For example, for levetiracetam (Keppra) oral solution (1 ml contains 100 mg of levetiracetam). The drug is indicated as monotherapy (drug of first choice) in the treatment of partial seizures with or without secondary generalization in adults and adolescents over 16 years of age with newly diagnosed epilepsy and as part of complex therapy: in the treatment of partial seizures with or without secondary generalization in adults and adolescents older than 1 month suffering from epilepsy; myoclonic seizures in adults and adolescents over 12 years of age suffering from juvenile myoclonic epilepsy; primary generalized convulsive (tonic-clonic) seizures in adults and adolescents over 12 years of age suffering from idiopathic generalized epilepsy.
Phenobarbital as a side effect causes sedation, in children - irritability, dysphoria, hyperkinetic disorders, decreased memory and intelligence, i.e., what are the main symptoms of the disorders in question. That is why treatment with phenobarbital is considered one of the factors in the development of epileptic psychoses, and the transition from it to valproate is the main condition for treatment [25]. The same is true for phenytoin. As for carbamazepine, it is widely used as a behavior corrector and anxiety disorders, in particular for attention deficit hyperactivity disorder, panic attacks, and also as a prophylactic for addiction disorders (it was on this basis that one of our patients received it).
The use of carbamazepine in epileptic encephalopathy as a first-choice drug is contraindicated, since the main task is to suppress epileptic brain activity, and carbamazepine often causes its intensification or appearance [26]. In this regard, the failure of treatment with finlepsin in sufficient doses of the patient, in whom both electroencephalographic and clinical improvement was achieved when switching to valproate, is characteristic, and the gradual withdrawal of finlepsin against the background of an already stabilized dose of depakine chrono was associated with further improvement in the EEG. This additional improvement associated with finlepsin withdrawal may be explained by the elimination of its facilitative effect on epileptiform discharges and its pharmacokinetic activation of liver enzymes that degrade valproate [5].
The duration of treatment and dose vary depending on the form of the disease and are determined empirically. If unsuccessful, topiramate, lamotrigine, and felbamate are used [32]. If ineffective, steroids are used in patients with speech disorders associated with Landau-Kleffner syndromes and electrical status in slow-wave sleep.
Antipsychotics and tricyclic antidepressants, nootropics, carbamazepine, phenytoin, and phenobarbital are contraindicated in this group of patients. Only if psychotic symptoms or depression persist, despite the complete suppression of epileptiform activity on the EEG (which was not the case in our observations), is it permissible to use antipsychotics of the group pimozide, sulpiride and antidepressants that block the reuptake of serotonin, such as Prozac (fluoxetine), fevarin (fluvoxamine ), to a lesser extent influencing the threshold of convulsive readiness of the brain. At the same time, intensive rehabilitation therapy should be carried out (with the participation of psychologists, speech therapists and other specialists) [30].
The prognosis of the disease depends on the etiology. In the absence of significant structural disorders of the brain and its congenital pathology, with proper treatment, favorable. With insufficiently persistent treatment aimed at suppressing epileptic activity on the EEG, the progression of psychocognitive disorders with the likelihood of severe social disorders. The addition of seizures and the occurrence of convulsive status is possible [28].
Conclusion: it should be emphasized once again that the main indicator of the correct choice of drug, dose and treatment success is the normalization of the EEG. Clinical improvement in most cases is manifested by a certain, often significant lag relative to EEG data. As has already been reported, another opportunity to improve the effectiveness and tolerability of antiepileptic therapy is the creation of new dosage forms of drugs that already exist in certain dosage forms, most often tablets.
Literature:
- Aggravation of epileptic seizures by anticonvulsants and the development of tolerance to them: scientific publication / A. Yu. Ermakov, S. R. Boldyreva, O. V. Gaponova, I. V. Shulyakova // Attending physician. - M., 2006. - No. 8 .- P.48–51.
- Vaskova L. Epilepsy: let's learn to count. Pharmacoeconomic aspects of the disease // Medical newspaper. - M., 2007. - No. 19 (March 16). - P. 12–13.
- Vertkin A.L., Moskvichev V.G., Skorikova Yu.S. Clinical comments on the standard of medical care for patients with epilepsy // Directory of paramedics and midwives. - M., 2008. - No. 10. - P.56–63.
- Dynamics of indicators as a criterion for assessing the quality of treatment of epilepsy with anticonvulsants: scientific publication / A. S. Shershever, S. A. Lavrova, A. V. Telegin, A. V. Grib // Attending physician. - M., 2006. - No. 10.- P.81–83.
- Dushanova T. A., Dikhanbaeva T. A., Karakulova A. A. Convulex is a valproic drug in the treatment of various forms of epilepsy. In: Current problems of neurology // Inter. conf. dedicated 70th birthday. Prof. M. Kh. Khafizova. 22–23 Oct. 2004 – Almaty, 2004. -S. 88–90.
- Ermolenko N. A., Ermakov A. Yu., Buchneva I. A. Treatment of epileptic encephalopathies with electrical status of slow-wave sleep // Journal "Medical Council". - 2008. - No. 3–4.
- Zenkov L. R., Prityko A. G. Drug-resistant epilepsies (manual for doctors). -M.: “Medpress inform”, 2003. -197 p.
- Karlov V. A. Epileptic encephalopathy. Journal of Neurology and Psychiatry 2006; 2:4–9.
- Kissin M. Ya. Clinical epileptology. M.: GEOTAR-Media. 2009. 256 p.
- Keppra in the treatment of epilepsy: effectiveness and tolerability / K. Yu. Mukhin, S. V. Pilia, V. A. Chadayev and others // Journal of Neurology and Psychiatry. - M., 2005. - No. 1. - P.49 –51.
- Likhterman B. New in the diagnosis and treatment of epilepsy. Fundamental sciences - clinical epileptology // Medical newspaper. - M., 2005. - No. 77 (Oct. 5). - P.7.
- Mono and polytherapy of epilepsy with the new drug convulsofin. // Medical Express. Central Asian medical journal for medical practitioners. -2002. -No. 1. (4). 473. - pp. 46–47.
- Mukhin K. Yu. Petrukhin A. S. Mironov M. B. Epileptic syndromes. Diagnostics and therapy. A reference guide for doctors. Moscow 2008. p57–77.
- Petrukhin A. S., Mukhin K. Yu., Kalinina L. V. Lamictal: poly- and monotherapy for epilepsy // Journal of Psychiatry and Psychopharmacotherapy. -2004. -No. 1. -S. 3–7.
- Shamansurov Sh. Sh., Gulyamova D. N. Neuroprotective effect of glycine in convulsive conditions in children. In the collection: Current problems of neurology // International conference, dedicated. 70th birthday. prof. M. Kh. Khafizova. 22–23 Oct. 2004 -Almaty, 2004. -S. 306–307.
- Abend NS, Dlugos DJ Nonconvulsive status epilepticus in a pediatric intensive care unit.// Pediatr. Neurol. 2007 V. 37 p. 165–170.
- Andrade-Machado R, Benjumea-Cuartas V, Jaramillo-Jimenez E. Lacosamide in lennox-gastaut syndrome: case report // Clin Neuropharmacol. 2012 May;35(3):148–9.
- Aydin OF, Senbil N., Gurer YK Nonconvulsive status epilepticus on EEG in a case with subacute sclerosing panencephalitis.// J. Child. Neurol/ 2006. V. 21 p. 256–260.
- Chien YH, Lin MI, Weng WC, Du JC, Lee WT. Dextromethorphan in the treatment of early myoclonic encephalopathy evolving into migrating partial seizures in infancy // J Formos Med Assoc. 2012 May;111(5):290–4. Epub 2012 May 14.
- Cross JH. The ketogenic diet in the treatment of Lennox-Gastaut syndrome // Dev Med Child Neurol. 2012 May;54(5):394–5. doi: 10.1111/j.1469–8749.2012.04276.x.
- Cusmai R, Martinelli D, Moavero R, Dionisi Vici C, Vigevano F, Castana C, Elia M, Bernabei S, Bevivino E. Ketogenic diet in early myoclonic encephalopathy due to non ketotic hyperglycinemia // Eur J Paediatr Neurol. 2012 Jan 17.
- de Haan TR, Bijleveld YA, van der Lee JH, Groenendaal F, van den Broek MP, Rademaker CM, van Straaten HL, van Weissenbruch MM Pharmacokinetics and pharmacodynamics of medication in asphyxiated newborns during controlled hypothermia. The PharmaCool multicenter study // BMC Pediatr. 2012 May 22;12:45.
- Fejerman N, Caraballo R, Cersósimo R, Ferraro SM, Galicchio S, Amartino H. Sulthiame add-on therapy in children with focal epilepsies associated with encephalopathy related to electrical status epilepticus during slow sleep (ESES). // Epilepsy. 2012 Apr 17. doi: 10.1111/j.1528–1167.2012.03458.x.
- Grimminck B, de Jong H, Sluzewski M, Obihara CC. A child with convulsions of unknown origin: posterior reversible encephalopathy syndrome // Ned Tijdschr Geneeskd. 2012;156(17):A3920.
- Lee EH, Yum MS, Jeong MH, Lee KY, Ko TS. A case of malignant migrating partial seizures in infancy as a continuum of infantile epileptic encephalopathy // Brain Dev. 2011 Dec 23.
- Lerman-Sagie T, Ben Zeev B, Kramer U. Immunoglobulin treatment for severe childhood epilepsy. // Pediatr Neurol. 2012 Jun;46(6):375–81.
- Li Y, Jenny D, Castaldo J. Posterior reversible encephalopathy syndrome: clinicoradiological spectrum and therapeutic strategies // Hosp Pract (Minneap). 2012 Feb;40(1):202–13.
- Pawar PS, Noviawaty I, Zaidat OO. Unusual case of intra-arterial doxorubicin chemoembolization-associated posterior reversible encephalopathy syndrome // Neurologist. 2012 Jan;18(1):49–50.
- Romem A, Galante O, Shelef I, Almog Y. Posterior reversible encephalopathy syndrome complicating septic shock.// Isr Med Assoc J. 2011 Dec;13(12):776–8.
- Thammongkol S, Vears DF, Bicknell-Royle J, Nation J, Draffin K, Stewart KG, Scheffer IE, Mackay MT. Efficacy of the ketogenic diet: which epilepsies respond? // Epilepsy. 2012 Mar;53(3):e55–9. doi: 10.1111/j.1528–1167.2011.03394.x.
- Veeramah KR, O'Brien JE, Meisler MH, Cheng X, Dib-Hajj SD, Waxman SG, Talwar D, Girirajan S, Eichler EE, Restifo LL, Erickson RP, Hammer MF. De novo pathogenic SCN8A mutation identified by whole-genome sequencing of a family quartet affected by infantile epileptic encephalopathy and SUDEP // Am J Hum Genet. 2012 Mar 9;90(3):502–10. Epub 2012 Feb 23.
- Writzl K, Primec ZR, Stražišar BG, Osredkar D. Early onset West syndrome with severe hypomyelination and coloboma-like optic discs in a girl with SPTAN1 mutation // Epilepsia. 2012 Jun;53(6):e106–10.