Эпилептическая энцефалопатия — прогрессирующее нарушение мозговых функций на фоне клинически проявленной или бессудорожной эпилепсии. Чаще обнаруживается в детском возрасте. У взрослых проявляется расстройствами аффекта, личностными изменениями, иногда – зависимостями.
предлагает профессиональную помощь при эпилепсии. Звоните по тел. 8(969)060-93-93.
Причины
В основе лежат органические изменения церебральных структур, обусловленные генетическими и приобретенными влияниями. Провоцирующими факторами у детей считаются:
- Неблагоприятная наследственность. Младенческая эпилептическая энцефалопатия обнаруживается при некоторых хромосомных аномалиях и генных мутациях.
- Сбои метаболизма. ЭЭ потенцируется пропионовой ацидемией, метилмалоновой ацидурией, некротической гипергицинемией и некоторыми другими врожденными метаболическими патологиями.
- Опухоли мозга. Риск возникновения болезни зависит от расположения и вида новообразования.
- Аномалии развития. Причиной становятся фокальные кортикальные дисплазии, церебральная дисгенезия, синдром Экарди, порэнцефалия, гемимегалэнцефалия.
- Поражения ЦНС. Болезнь выявляется при повреждениях мозга во внутриутробном, младенческом и раннем детском возрасте.
В число других неблагоприятных обстоятельств у всех категорий больных входят патологии беременности, недоношенность, психические расстройства и химические зависимости у родителей, эпилептоидные синдромы у родственников, легкие ЧМТ и фебрильные судороги.
СОВРЕМЕННЫЕ АСПЕКТЫ ГЕНЕТИКИ ЭПИЛЕПТИЧЕСКОЙ ЭНЦЕФАЛОПАТИИ И СИМПТОМАТИЧЕСКОЙ ЭПИЛЕПСИИ У ДЕТЕЙ
Эпилепсия – одна из наиболее актуальных проблем в педиатрической неврологии. Распространенность эпилепсии в популяции достаточно высока и составляет 0,5-0,75%, а среди детского населения достигает 1%. Примерно 7% детей переносят хотя бы один приступ в течение жизни. [1, 3].
По данным Алимовой В.С. (2005), которая изучила частоту и распространенность эпилепсии среди детей, подростков и взрослого населения Республики Узбекистан за период 2001-2003 года выявлено, что в 2003 году больные эпилепсией составили 5,45% от всех неврологических больных. При этом частота заболеваемости эпилепсией колебалась от 1,52 на 1000 населения в 2001 году до 1,12 в 2003 году. Автор отмечает, что за изучаемый период заболеваемость среди детей и подростков значительно превысила эти показатели среди взрослого населения. [2,5].
С генетической точки зрения, все формы эпилепсии можно подразделить на моногенные заболевания и заболевания с наследственной предрасположенностью. Согласно последним достижениям молекулярно-генетических исследований различных форм эпилепсии выделяют несколько групп генов, участвующих в развитии идиопатической эпилепсии [8, 10]:
- гены потенциал — зависимых ионных каналов, к которым относят, прежде всего, гены натриевого, калиевого, кальциевого и хлорного каналов.
Последние достижения молекулярно-генетических исследований эпилепсии выявили, что в основе некоторых форм эпилепсии лежит нарушение деятельности именно ионных каналов. Вполне вероятно, что их широкое распространение в головном мозге, а также мутации в них способствуют возникновению судорожной активности. Нарушения в этих генах могут прямым или косвенным образом приводить к аномальному повышению возбудимости нейронов. [6, 7].
- гены рецепторов и переносчиков нейромедиаторов торможения и возбуждения (гамма-аминомасляной кислоты, серотонина, глутамата).
Несмотря на то, что в последние годы во всем мире ведутся активные молекулярно-генетические исследования эпилепсии и уже известно более десятка генов, мутации в которых приводят к развитию разных форм эпилепсии, в этой области все еще остается много неясного, как и в эпилептогенезе в целом [14,16].
Таким образом, раскрытие генетических аспектов эпилепсии необходимо для четкого понимания этиологии и патогенеза этого заболевания, поиска новых путей коррекции. Однако конкретная роль генетических факторов эпилепсии остается неизвестной, особенно среди детей узбекской национальности.
В целом, в последние годы, с использованием молекулярных технологий достигнут очевидный существенный прогресс в прояснении молекулярно-генетических основ эпилепсии. Молекулярная дифференциация случаев заболевания обеспечивает клиницистов новыми диагностическими инструментами надежной диагностики, позволяющей точно оценить генетический риск, давать прогноз при консультировании и планировать рациональные терапевтические подходы.
Для выявления мутаций и полиморфизмов у изучаемых больных эпилептической энцефалопатии нами проведен анализ гена а 1 субъединицы нейронального натриевого канала (SCN1A), в которых ранее были выявлены мутации и полиморфизмы у больных с различными формами эпилепсии.
Ген SCN1A, кодирующий al- субъединицу нейронального натриевого канала, охватывает около 70 000 пар оснований (п.о.) геномной ДНК, включает 26 экзонов (длиной от 8 п.о. до 3260 п.о.) и расположенных между ними интронов [18]. Продукт гена SCN1A состоит из четырех гомологичных доменов, содержащих шесть трансмембранных сегментов и соединенных внутриклеточными петлями. Уникальная последовательность сегмента S4 несет положительный остаток в каждой третьей позиции трансмембранного сегмента. Предполагается, что это свойство обеспечивает чувствительность канала к электрическому полю. Структура, формирующая ионную пору, располагается между высоко-консервативными S5 и S6 сегментами [19].
Ген SCN1A принадлежит к семейству генов, которые обеспечивают инструкции для натриевых каналов. Эти каналы, которые транспортируют положительно заряженные атомы натрия (ионы натрия) в клетки, играют ключевую роль в способности клетки генерировать и передавать электрические сигналы.
Ген SCN1A содержит инструкции для создания одной части (альфа-субъединицы) из натриевых каналов называется NaV1.1. Эти каналы находятся в головном мозге и мышцах, где они контролируют поток ионов натрия в клетку. В мозге NaV1.1 каналов, участвующих в передаче сигналов от одной нервной клетки (нейрона) на другую. Связь между нейронами зависит от химических веществ, называемых нейротрансмиттеров, которые освобождены от одного нейрона и подхватили соседние нейроны. Поток ионов натрия через каналы NaV1.1 помогает определить, когда медиаторы будут освобождены.
Более 150 мутаций в гене SCN1A были связаны с различными нарушениями захвата, которые начинаются в младенчестве или детстве. Некоторые из этих условий относительно мягким. Эти условия включают в себя простые фебрильные (лихорадка связанных) припадки, которые начинаются в младенчестве и обычно прекращаются в возрасте 5 и генерализованной эпилепсии с фебрильных судорог плюс (GEFS +). GEFS + включает в лихорадочных и других видов припадков, которые могут сохраняться после детства. Другие условия вызывают более серьезные приступы, которые длятся дольше и может быть трудно контролировать. Эти повторяющиеся приступы могут ухудшаться с течением времени и приводит к снижению функции мозга. Тяжелые нарушения захвата вызваны мутациями SCN1A включают тяжелые миоклонические эпилепсии младенчества (SMEI) и неразрешимыми эпилепсия детства с генерализованными тонико-клоническими судорогами (ICE-GTC). [12].
SCN1A мутации, лежащие в основе эпилептических припадков имеют различные эффекты на функцию канала NaV1.1.Мягкие расстройства вызываются мутациями, которые изменяют одной аминокислоты в канале, которые изменяют структуру канала. Более серьезные нарушения захвата может быть результатом различных изменений в гене SCN1A. Некоторые мутации приводят к производству нефункциональные версии канала NaV1.1 или уменьшить количество этих каналов производится в каждой ячейке. Другие мутации изменить одну аминокислот в критических областях канала. Все эти генетические изменения влияют на способность NaV1.1 каналы для транспортировки ионов натрия в нейронах. Неясно, однако, почему эти генетические изменения приводят к такой большой диапазон приступов.
Общее изменение (полиморфизмом) гена SCN1A была связана с эффективностью некоторых анти-захвата лекарства. Этот полиморфизм, который записывается в виде ICS5N +5 G> A, изменяется один строительный блок ДНК (нуклеотид) в гене SCN1A. Исследования показывают, что этот полиморфизм связан с максимально безопасного количества (дозы) о борьбе с захватом наркотиков фенитоин и карбамазепин. Эти препараты лечения эпилепсии, блокируя натриевые каналы (например, NaV1.1) в нейронах.Доза, которая слишком мала, не может эффективно контролировать припадки, а доза, слишком большой может вызвать нежелательные побочные эффекты. Сейчас врачи должны использовать проб и ошибок, чтобы определить правильную дозу для каждого человека. Исследователи надеются, что врачи в один прекрасный день смогут использовать ICS5N +5 G> A полиморфизма для определения самых безопасных и эффективных доз анти-захвата лекарства для каждого человека.
Цитогенетическое расположение SCN1A гена — 2q24.3
Молекулярная Расположение на хромосоме 2: 166 845 669 пар оснований, чтобы 167 005 641 (рис. 1)
Рис. 1. Ген SCN1A расположен на длинном (д) плече хромосомы 2 в позиции 24.3.
Точнее, ген SCN1A находится от 166 845 669 пар оснований, до 167 005 641 пар оснований, на хромосоме 2.
В настоящее время известно более 70 мутаций в гене SCN1A, вызывающих развитие таких форм эпилепсии, как доброкачественные семейные судороги новорожденных, генерализованная эпилепся с фебрильными судорогами плюс и фебрильные судороги [14].
Мутации, приводящие к развитию доброкачественных семейных судорог раннего детства, являются нонсенс — мутациями, мутациями сайтов сплай-синга, сдвига рамки считывания и, в основном, встречаются в S5-S6 сегментах и петле между ними. Чаще всего они полностью нарушают функцию нейронального натриевого канала [5].
Более «мягкий» фенотип генерализованных фебрильных судорог плюс (GEFS+) обуславливают миссенс мутации в петле, соединяющей S2 и S3 сегменты гена SCN1A, ведущие к увеличению времени восстановления активности канала после инактивации [8,11]
Из всей выборки больных эпилептической энцефалопатии у детей (n=20) полиморфизм 3184 A–G встречался в гомозиготном состоянии (3184*А/*А) у 8 больных (40%), в гетерозиготном состоянии (3184*A/*G) — у 11 (56%), а гомозиготный генотип (3184*G/*G) был определен только у 1 пациента (4%) (рис. 2).
Рис. 2. Частота встречаемости полиморфизма 3184 A–G у детей с эпилептической энцефалопатии
Так нами было установлено, что для эпилептической энцефалопатии картирован ряд локусов сцепления: 40% — 168bp; 56% — 168, 145 и 23bp и 4% -145 и 23 bp (рис.3).
Рис. 3. Картированый ряд локусов сцепления у детей с эпилептической энцефалопатией узбекской национальности
Как видно из полученных данных у большинства больных с эпилептической энцефалопатией обнаружены мутации в гене SCN1A, кодирующем а1 субъединицу нейронального натриевого канала, что обуславливает увеличение времени восстановления активности канала после интактивации, что в свою очередь также приводит к гипервозбудимости нейрона.
В процессе развития данной формы заболевания предполагается участие нескольких взаимодействующих между собой генов, расположенных в данных областях. Вклад каждого из них в риск развития эпилептической энцефалопатии может быть небольшим, но, тем не менее, приводящим к наследуемым изменениям, предрасполагающим к судорожной активности.
Таким образом, учитывая все вышеизложенное можно сделать следующее заключение мутации и полиморфизмы в гене SCN1A нейронального натриевого канала приводят к увеличению времени восстановления активности канала после инактивации и, как следствие, провоцируют гипервозбудимость нейронов. Обнаруженные нами изменения нуклеотидной последовательности гена SCN1A могут вносить определенный вклад в развитие предрасположенности к эпилептической энцефалопатии у детей узбекской национальности.
- Akre B., Rasmussen M; Lundby R. Pyridoxine – dependent seizures //Norwegian Journal In formation. -2004. -Р. 162-164.
- Alkan A., Kutlu R., Aslan M. Pyridoxine – Dependent Seizures: Magnetic Resonance Spectroscopy Findings // Journal of Child Neurology. -2004. Vol. 19, №1. -Р
- Battaglioli G., Rosen D. R., Gospe S. M. Jr., Glutamate decarboxylase is not denetically linked to pyridoxine-dependent seizures //Neurology. -2000. -№25. 55 (2). -309-311.
- Berkovic S., Howell R., Hay D. Epilepsies in twins: genetics of the major epilepsy syndromes II Neurol. — 1998. — V. 43. — P. 435-445.
- Biervert C, Schroeder В., Kubisch C, et al. A potassium channel mutation in neonatal human epilepsy II Science — 1998. — 279. — P. 403-409.
- Caraballo R., Garro F., Cersosimo R. Pyridoxine dependence: the importance of clinical diagnosis and early treatment //Revista de neurologia—2004. -№38. -P. 49-52.
- Jang Y. M., Kim D. W., Kang T. C. Human pyridoxal phosphatase. Molecular cloning, functional expression, and tissue distribution // Journal of biological chemistry. -2003. -№12. -Р. 5040-5046.
- Kaelin A., Caser J. P., Jaeger R. Vitamin B6 metabolites in idiopatic calcium stone formers: no evidence for a link to hyperoxaluria // Urological research. -2004. -№32 (1). -P. 61-68.
- Kannan K., Jain S. K. Effect of vitamin B6 on oxygen radicals, mitochondrial membrane potential, and lipid peroxidation in H2 O2 treated U 937 monocites // Free radical biology medicine. -2004. -№15;36 (4). -P. 423-428.
- Kholin A.A., Lemeshko I.D., Il’ina E.S, Voronkova K.V., Petrukhin A.S. Seizure characteristics of early myoclonic encephalopathy // Extended Abstracts. 2nd East Mediterranean Epilepsy Congress. Dubai.-2010.- A-246-0004-00036.
- Kholin A.A., Voronkova K., Aivazjan S., Shevchenko A., Lemeshko I., Il’ina E.S., Petrukhin A.S. Features of status epilepticus at infancy and early childhood // 28th International Epilepsy Congress 28th June-2nd July 2009.- P.757.
- Maeda T., Inutsuka M., Goto K., Irumi T. Transient nonketotik hyperglycinemia in an asphyxiated patient with pyridoxine-dependent seizures // Pediatr. Neurol. — -№22 (2). -P. 225-227.
- Najafi Reza Mohammad, Tamizifar Babak. Drug discontinuation in epileptic children: predictive value of the EEG // Archives of Iranian Medicine. -2002. -Vol. 5, № 3, — P. 157-161.
- Okuda T., Sumiya T., Iwai N., Viyata T. Piridoxine-5-phosphate oxidase is a candidate gene responsible for hypertension in Dohl-s rats //Biochemical and biophysical research communications. -2004. -№313 (3). -P. 647-653.
- Said H. M. Recent advances in carrier-mediated intestinal absorption of water-soluble vitamins //Annual review of physiology. -2004. -№66. -P. 419-446.
- Schulre-Bonhage A., Kurthen M., Walger P. Pharmacorefractory status epilepticus due to low vitamin B6 levels during pregnancy //J. Epilepsia. -2004. -№45(1). -P.81-84.
- Tsuge H., Hotta N., Hayakawa T. Effects of vitamin B6 on (n-3) polyunsaturated fatty acid metabolism //J. Nutr. -2000. -№130. -P. 333-334.
- Voronkova K., Kholin A.A., Lemeshko I., Il’ina E.S., Petrukhin A.S. Seizure characteristics of Dravet Syndrome // 28th International Epilepsy Congress 28th June-2nd July 2009.- P.750.
- Wanden L. R. Magnesium absorbtion using stable isotope tracers in healthy children and children treated for leukemia //Nutrition. -2001. -№3. -P. 221-224.
Классификация
С учетом времени возникновения, особенностей течения и преобладающей симптоматики различают два вида ЭЭ:
- Первый. Ранняя эпилептическая энцефалопатия. Течение тяжелое, превалируют усугубляющиеся нарушения речи, интеллекта, познавательных способностей. В группу входят синдромы Леннокса и Отахары, детский спазм и пр.
- Второй. Чаще диагностируется у взрослых. Пароксизмы отсутствуют, эпи-активность выявляется только по данным дополнительных исследований. Наблюдаются поведенческие, когнитивные, психические и социальные нарушения.
Этиологические факторы
Причины развития ранней эпилептической энцефалопатии разнообразны. Их можно разделить на генетические и негенетические. К первой группе относятся:
- пороки развития и перинатальные заболевания, которые вызывают структурное повреждение головного мозга;
- заболевания, которые сопровождаются нарушением обмена веществ (митохондриальные болезни, недостаток фермента сульфатоксидазы, фенилкетонурия и др.);
- другие состояния, при которых возникает поражение структур головного мозга (инсульт, кортикальная дисплазия, травмы, туберозный склероз, инфекционные заболевания).
К генетическим причинам эпилептической энцефалопатии относятся мутации в генах, которых насчитывается более 35: STXBP1, KCNQ2, CASK, SCN2A, SLC25A22, STK9, PLCB1 и т. д. Также заболевание может сочетаться с такими хромосомными аномалиями, как синдром Дауна или синдром Ангельмана. По типу наследования эпилептические энцефалопатии могут быть Х-сцепленными доминантными и рецессивными, а также аутосомно-доминантными и аутосомно-рецессивными.
Симптомы у детей
Перечень осложнений первого рода включает изменения психики и поведения, нейропсихологические расстройства, характерные для органических заболеваний ЦНС. У грудничков выявляется частое срыгивание, проблемы при грудном вскармливании, плохой сон, повышение мышечного тонуса, перебои в работе сердца. Возможны неадекватные реакции на внешние раздражители (звуки, свет, изменение положения тела), беспокойство, капризы, частый плач.
У дошкольников сохраняется повышенный мышечный тонус, проблемы со сном, обмороки и сильные головные боли. В эмоциональной сфере отмечаются частые приступы гнева, неустойчивость настроения, неуверенность, в когнитивной — сложности при переключении внимания, плохое запоминание. Начинается формирование эпилептоидной ригидности.
У детей школьного возраста на первый план выходят расстройства познавательной деятельности. Пациенты плохо запоминают материал, не справляются с заданиями, тяжело адаптируются к новым правилам и обстановке. Сохраняются головокружения, головные боли, обморочные состояния. Завершается формирование эпилептоидного типа личности.
ЭЭ становится причиной комплексных нарушений психики, речевой сферы, физического развития и двигательных навыков. При прогрессирующем течении возможны гидроцефалия, олигофрения, слабоумие, депрессии, мании, психозы. Неспособность к обучению оборачивается социальной дезадаптацией, невозможностью получения профессии, потребностью в постороннем уходе. При своевременном начале лечения ЭЭ в достаточной степени компенсируется, дети оканчивают школу, работают, создают семьи.
Диагностика
Производится неврологом, предполагает проведение комплексного обследования с использованием следующих методов:
- Неврологический осмотр. Включает исследование рефлексов, мышечной силы, различных видов чувствительности.
- Нейровизуализация. На электроэнцефалограммах видны участки эпиактивности. На МРТ могут просматриваться признаки структурных изменений. По данным дуплексного сканирования и допплерографии оценивается мозговое кровообращение.
- Консультация психиатра. Предполагает изучение характериологических, когнитивных, поведенческих особенностей, уточнение тяжести психических нарушений.
- Психологические тесты. Включают опросники, проективные методы, патопсихологическое обследование.
- Лабораторные анализы. Информативны при некоторых наследственных и метаболических вариантах расстройства.
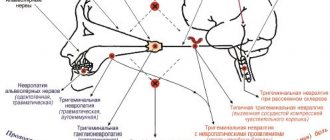
Характер психических и неврологических проявлений коррелирует с расположением эпиочага на ЭЭГ. При локализации в левом полушарии превалируют расстройства речи, письма и счета, в правом — дезартикуляция, неспособность регулировать и распознавать изменения тона, ритма и интонации в разговоре, трудности при определении расположения и размера объектов. При поражении срединных структур отмечаются эксцентричное, асоциальное или агрессивное поведение, аутизм.
При отсутствии приступов определить, что это у взрослых, бывает непросто. В пользу эпилептической природы энцефалопатии свидетельствует совпадение расположения очага с нарушениями определенных психических функций, связь между эпиактивностью и появлением симптомов, улучшение состояния при приеме противоэпилептических препаратов.
Стоимость услуг
| КОНСУЛЬТАЦИИ СПЕЦИАЛИСТОВ | |
| Первичная консультация врача психиатра (60 мин.) | 6 000 руб. |
| Повторная консультация | 5 000 руб. |
| Консультация психиатра-нарколога (60 мин.) | 5 000 руб. |
| Консультация психолога | 3 500 руб. |
| Консультация Громовой Е.В. (50 минут) | 12 000 руб. |
| ПСИХОТЕРАПИЯ | |
| Психотерапия (сеанс) | 7 000 руб. |
| Психотерапия (5 сеансов) | 30 000 руб. |
| Психотерапия (10 сеансов) | 60 000 руб. |
| Групповая психотерапия (3-7 человек) | 3 500 руб. |
| Сеанс психотерапии у Громовой Е.В. (50 минут) | 12 000 руб. |
| ЛЕЧЕНИЕ В СТАЦИОНАРЕ | |
| Палата 4-х местное размещение | 10 000 руб./сутки |
| Палата 3-х местное размещение | 13 000 руб./сутки |
| Палата 1 местное размещение VIP | 23 000 руб./сутки |
| Индивидуальный пост | 5 000 руб. |
| ПИТ | 15 000 руб./сутки |
В данном перечне представлены не все цены на услуги, которые предоставляет наша клиника. С полным прайсом можно ознакомиться на странице «Цены», либо по телефону: 8(969)060-93-93. Первичная консультация БЕСПЛАТНА!
Лечение
Программа лечения эпилептических энцефалопатий составляется индивидуально с учетом возраста, разновидности и причин возникновения болезни. В рамках терапии ЭЭ применяются следующие лекарственные средства:
- Противосудорожные. Расслабляют мышцы, устраняют судорожные приступы. В качестве препаратов первой линии обычно используют вальпроевую кислоту.
- Транквилизаторы. Показаны больным с малыми припадками.
- Седативные. Помогают стабилизировать эмоциональное состояние.
- Сосудорасширяющие. Улучшают кровоток в церебральных структурах.
- Ноотропы. Повышают активность мозговых клеток, способствуют развитию памяти, внимания.
- Витамины, аминокислоты. Стимулируют обменные процессы в нервной ткани.
Дополнительно применяют немедикаментозные методы. Детям показаны психокоррекционные занятия, работа с логопедом. Всем пациентам полезны ЛФК, массаж. Рекомендована нормализация режима дня, продуманное чередование нагрузок и периодов отдыха. Прогноз определяется формой болезни, своевременностью и адекватностью медицинской помощи.
Записаться на прием к неврологу в клинике «Лето» можно по тел. 8(969)060-93-93.
Современные принципы лечения эпилептической энцефалопатии у детей
В статье представлен обзор литературы современных принципов лечения эпилептической энцефалопатии. Основным показателем правильности выбора препарата, дозы и успешности лечения является нормализация нейрофизиологических данных. Клиническое улучшение в большинстве случаев проявляется определенным, нередко значительным отставанием относительно данных электроэнцефалографии. Как уже сообщалось, еще одна возможность для улучшения показателей эффективности и переносимости антиэпилептической терапии — создание новых лекарственных форм препаратов, уже существующих в определенных лекарственных формах, чаще всего — в таблетках.
The article presents a review of the literature of modern principles of treatment of epileptic encephalopathy. The main indicator of the correctness of the choice of drug, dosage and treatment success is normalization of neurophysiological data. Clinical improvement in most cases manifested a certain, often considerable delay relative to the data of electroencephalography. As previously reported, another opportunity for improvements in the efficacy and tolerability of antiepileptic therapy — development of new dosage forms of drugs that already exist in certain dosage forms, more often — in the tablets.
Поиск возможностей для увеличения эффективности антиэпилептических препаратов (АЭП) включает: синтез новых лекарственных формул или модификацию уже синтезированных молекул, определение новых показаний к назначению, поиск новых лекарственных форм, новых рациональных комбинаций препаратов и другие. Современные методы решения проблемы побочных эффектов АЭП включают: профилактику (назначение АЭП с учетом общего соматического и психического здоровья пациента и его ближайших родственников), коррекцию побочных эффектов, мониторинг состояния: своевременные обследования, консультации специалистов [7. 29].
Положительное влияние у большинства пациентов противоэпилептической терапии, направленной на подавление эпилептической активности на ЭЭГ, а также тесная временная связь между регрессом такой эпилептической активности и клинической симптоматики подтверждают ключевую роль эпилептической мозговой активности в развитии устойчивой психиатрической и поведенческой симптоматики [4].
Отмечаемые в литературе [6] безуспешность применения нейролептиков и антидепрессантов при психозах, связанных с эпилептическими разрядами в лимбико-ретикулярной системе, как и эффективность при них противоэпилептических средств, определяются у 80 % больных, что соответствует и нашим данным. Снижая порог судорожной готовности и соответственно активируя эпилептическую активность, антидепрессанты и нейролептики не только не дают эффекта, но даже, как это было у наших пациентов, усугубляют симптоматику [12].
Следует отметить, что значительно быстрее — обычно на 3–5-й день при стабильной терапевтической дозе противоэпилептического препарата, регрессируют собственно психические нарушения (тревога, агрессивность, дезориентировка, расстройства памяти и мышления), что, по-видимому, определяется нормализацией нейрональных систем лимбико-ретикулярного комплекса после прекращения эпилептической активности. Восстановление речи, познавательных процессов, социальная адаптация происходят постепенно в течение недель, месяцев и лет, поскольку эти навыки требуют научения в процессе повседневной жизни [14].
Препаратом первого выбора при лечении эпилептических непароксизмальных расстройств и энцефалопатий является вальпроевая кислота (депакин хроно, депакин хроносфера) [2, 10, 18, 23]. Она подавляет эпилептическую активность в мозге. Нормализация ЭЭГ начинается уже на субтерапевтических дозах, что позволяет на 3–5-й день подбора терапевтической дозы убедиться в правильности выбора препарата, а в дальнейшем по динамике ЭЭГ корректировать дозу и контролировать успешность лечения [15]. Нормализация в ЭЭГ коррелирует с клиническим улучшением, что особенно важно, поскольку у пациентов нет припадков, по динамике которых возможно было бы отслеживать эффективность, а клиническая нормализация психической и поведенческой симптоматики обычно отстает от нормализации ЭЭГ [3. 21]. Помимо противоэпилептического действия вальпроевая кислота обладает собственным противопсихотическим действием.
В США вальпроевая кислота составляет 35 % всех назначаемых при шизофрении препаратов [22]. Основным механизмом действия вальпроевой кислоты является увеличение ГАМКергического торможения. Подавляя избыточную разрядную активность в мозге, она предотвращает эксайтотоксическую гибель нейронов, что является одним из механизмов ее нейропротекторного действия. Нормализующее действие в отношении киназ, генной экспрессии и транскрипции приводит к нормализации пластических нейрональных процессов, что способствует прерыванию эпилептического процесса и выздоровлению пациента [20].
В приложении к лечению детских и младенческих эпилептических энцефалопатий, в основе которых лежит незрелость мозга, важно то, что вальпроевая кислота активирует рост дендритов и аксонов у нейробластов и образование из них хорошо организованной нейронной сети, благоприятствуя тем самым созреванию функциональных систем [13]. Эти данные хорошо согласуются с ускоренным созреванием параметров ЭЭГ, наблюдаемым при лечении непароксизмальных эпилептических расстройств вальпроевой кислотой [27]. Условием эффективного лечения эпилептических энцефалопатий является стабильность подавления эпилептиформной активности, достигаемая постоянством поддержания эффективной концентрации препарата в плазме.
Fejerman N. et al. [23] на основании обобщения материала четырех клиник пришли к выводу, что леветирацетам является наиболее эффективным препаратом первого выбора при этой форме. Соответственно, показана эффективность леветирацетама в лечении эпилептических расстройств речи, поведения, памяти и психики, общих расстройств развития у пациентов без припадков [31]. В эксперименте показана способность леветирацетама противодействовать киндлингу в амигдале, что определяет его потенциальную эффективность в препятствовании прогрессированию эпилептической энцефалопатии [16].
Ламотриджин (ламиктал) рекомендуется для лечения младенческих и детских катастрофических эпилептических синдромов: Уэста, Леннокса-Гасто, с постоянными комплексами спайк-волна в медленноволновом сне и других непароксизмальных эпилептических расстройств. Применение его при непароксизмальных эпилептических расстройствах и эпилептических энцефалопатиях определяется тем, что он подавляет эпилептиформную активность в ЭЭГ [17]. Ограничения в отношении ламотриджина в качестве препарата первого выбора при лечении эпилептических энцефалопатий определяются медленным титрованием, так что терапевтическая концентрация достигается спустя недели от начала лечения, а также тем, что он в некоторых случаях может утяжелять припадки, эпилептиформную активность в ЭЭГ и когнитивные и поведенческие нарушения [1].
Эффективность топирамата показана при катастрофических эпилептических энцефалопатиях — синдроме Леннокса — Гасто и Уэста. Данные об успешном лечении эпилепсии с электрическим эпилептическим статусом в медленноволновом сне говорят о возможности его применения при когнитивных и психических расстройствах, обусловленных этим состоянием. Есть также единичные сообщения о применении его при эпилептических расстройствах с аутистической регрессией и нарушении способности к обучению [9]. Вместе с тем основным побочным эффектом топирамата является его негативное влияние на психические, поведенческие и когнитивные функции у некоторых пациентов, что требует осторожного отношения к возможности его применения при психических и поведенческих эпилептических расстройствах, особенно у пациентов с нарушением вербальных функций, которое он сам может вызывать [10].
В качестве дополнительной терапии эпилептических энцефалопатий в резистентных к монотерапии случаях применяются бензодиазепины (клоназепам, клобазам, диазепам), этосуксимид (Суксилеп) [12]. При тяжелых формах эпилептических энцефалопатий и непароксизмальных психических, поведенческих и нейропсихологических эпилептических расстройств (синдромы Уэста, Леннокса — Гасто, Ландау — Клеффнера, эпилепсия с постоянными комплексами спайк-волна во сне, оперкулярный эпилептический статус, эпилептическая речевая диспраксия) применяется гормональная терапия, чаще всего препаратами адренокортикотропного гормона (АКТГ). Используют как натуральный АКТГ и его синтетический аналог Синактен депо, так и преднизолон. Дозировки препарата широко варьируют, поскольку индивидуальные пациенты отвечают на разные режимы лечения [8, 19].
На фармацевтическом рынке широко представлены новые формулы АЭП с новыми механизмами действия.
В России в 2010 был зарегистрирован лакосамид (вимпат) для лечения фокальных эпилепсий у пациентов старше 16 лет в комбинированной терапии. У лакосамида открыт новый противоэпилептический механизм. Препарат избирательно усиливает медленную инактивацию натриевых каналов без влияния на быструю инактивацию, в результате чего происходит: стабилизация гипервозбудимых нейрональных мембран, нормализация порогов активации и ингибирование повторяющихся нейрональных разрядов. В настоящее время представлена как таблетированная форма лакосамида, так и внутривенный раствор, который является альтернативой для пациентов, временно неспособных принимать препарат per os [11].
В настоящее время на стадии регистрации находятся и другие новейшие противоэпилептические препараты: зонисамид (зонегран) в капсулах в качестве дополнительного лекарственного препарата у взрослых при лечении парциальных эпилептических припадков с и без вторичной генерализации и руфинамид (иновелон) в таблетках в качестве дополнительного лекарственного препарата при лечении эпилептических припадков, ассоциированных с синдромом Леннокса — Гасто у пациентов старше 4 лет. У зонисамида механизм действия — комплексный, однако до конца не раскрыт. Предполагается, что он воздействует на потенциалзависимые натриевые и кальциевые каналы, а также снижает активность нейронов посредством гамма-аминомасляной кислоты.
Руфинамид является модулятором натиевых каналов. В США был зарегистрирован в 2004г., в Европе — 2007г. В клинических исследованиях убедительно доказана его эффективность в терапии дропатак (уменьшает их на 42,5 %, а эффект становится заметным уже в первые 2 недели). Недавно было показано, что руфинамид может быть эффективным в комплексной терапии эпилептических энцефалопатий и эпилептических синдромов, помимо синдрома Леннокса — Гасто, — синдрома Драве, злокачественных мигрирующих парциальных приступах младенчества и других [24].
Утверждены новые показания к назначению отдельных АЭП. Например для леветирацетама (кеппры) раствора для приема внутрь (1 мл содержит 100 мг леветирацетама). Препарат показан в качестве монотерапии (препарат первого выбора) при лечении парциальных припадков с вторичной генерализацией или без таковой у взрослых и подростков старше 16 лет с вновь диагностированной эпилепсией и в составе комплексной терапии: при лечении парциальных припадков с вторичной генерализацией или без таковой у взрослых и подростков старше 1 месяца, страдающих эпилепсией; миоклонических судорог у взрослых и подростков старше 12 лет, страдающих ювенильной миоклонической эпилепсией; первично-генерализованных судорожных (тонико-клонических) припадков у взрослых и подростков старше 12 лет, страдающих идиопатической генерализованной эпилепсией.
Фенобарбитал в качестве побочных результатов вызывает седативный эффект, у детей — раздражительность, дисфорию, гиперкинетические расстройства, снижение памяти и интеллекта, т. е. то, что является основными симптомами рассматриваемых расстройств. Именно поэтому одним из факторов развития эпилептических психозов считается лечение фенобарбиталом, а переход с него на вальпроат — главным условием лечения [25]. Сказанное справедливо и в отношении фенитоина. Что касается карбамазепина, то он широко применяется в качестве корректора поведения и тревожных расстройств, в частности при синдроме дефицита внимания и гиперактивности, при панических атаках, а также как профилактическое средство при расстройствах зависимости (именно на этом основании его получала одна из наших пациенток).
Применение карбамазепина при эпилептической энцефалопатии в качестве препарата первого выбора противопоказанным, поскольку основная задача заключается в подавлении эпилептической активности мозга, а карбамазепин нередко вызывает ее усиление или появление [26]. В этом плане характерна безуспешность лечения финлепсином в достаточных дозах пациентки, у которой как электроэнцефалографическое, так и клиническое улучшение было достигнуто при переходе на вальпроат, причем постепенная отмена финлепсина на фоне уже стабилизированной дозы депакина хроно ассоциировалась с дальнейшим улучшением ЭЭГ. Это дополнительное улучшение, связанное с отменой финлепсина, может объясняться устранением его облегчающего действия в отношении эпилептиформных разрядов и его фрмакокинетической активацией энзимов печени, разрушающих вальпроат [5].
Продолжительность лечения и дозы варьируют в зависимости от формы заболевания и определяются эмпирически. При безуспешности применяют топирамат, ламотриджин, фелбамат [32]. В случае неэффективности у пациентов с расстройствами речи, связанными с синдромами Ландау — Клеффнера и “электрическим статусом в медленноволновом сне” применяют стероиды.
Данной группе пациентов противопоказаны нейролептики и трициклические антидепрессанты, ноотропы, карбамазепин, фенитоин, фенобарбитал. Только при сохранении психотической симптоматики или депрессии, несмотря на полное подавление эпилептиформной активности на ЭЭГ (чего в наших наблюдениях не было), допустимо применение нейролептиков группы пимозида, сульпирида и антидепрессантов, блокирующих обратный захват серотонина, таких как прозак (флюоксетин), феварин (флювоксамин), в меньшей степени влияющих на порог судорожной готовности мозга. Одновременно должна проводиться интенсивная реабилитационная терапия (с участием психологов, логопедов и других специалистов) [30].
Прогноз заболевания зависит от этиологии. В отсутствие значительных структурных нарушений мозга и его врожденной патологии при правильном лечении благоприятный. При недостаточно настойчивом лечении, направленном на подавление эпилептической активности на ЭЭГ, — прогрессирование психокогнитивных нарушений с вероятностью тяжелых социальных расстройств. Возможно присоединение припадков и возникновение судорожного статуса [28].
Заключение: еще раз следует подчеркнуть, что основным показателем правильности выбора препарата, дозы и успешности лечения является нормализация ЭЭГ. Клиническое улучшение в большинстве случаев проявляется определенным, нередко значительным отставанием относительно данных ЭЭГ. Как уже сообщалось, еще одна возможность для улучшения показателей эффективности и переносимости антиэпилептической терапии — создание новых лекарственных форм препаратов, уже существующих в определенных лекарственных формах, чаще всего — в таблетках.
Литература:
- Аггравация эпилептических приступов антиконвульсантами и развитие толерантности к ним: научное издание/ А. Ю. Ермаков, С. Р. Болдырева, О. В. Гапонова, И. В. Шулякова// Лечащий врач.- М., 2006.- № 8.- С.48–51.
- Васькова Л. Эпилепсия: будем учиться считать. Фармакоэкономические аспекты заболевания// Медицинская газета.- М., 2007.- № 19 (16 марта).- С.12–13.
- Верткин А. Л., Москвичев В. Г., Скорикова Ю. С. Клинические комментарии к стандарту медицинской помощи больным с эпилепсией// Справочник фельдшера и акушерки.- М., 2008.- № 10.- С.56–63.
- Динамика показателей как критерий оценки качества лечения эпилепсии противосудорожными препаратами: научное издание/ А. С. Шершевер, С. А. Лаврова, А. В. Телегин, А. В. Гриб// Лечащий врач.- М., 2006.- № 10.- С.81–83.
- Душанова Т. А., Диханбаева Т. А., Каракулова А. А. Конвулекс — препарат вальпроевого ряда в терапии различных форм эпилепсии. В сб.: Актуальные проблемы неврологии //Меж. конф. посв. 70-летию со дня рожд. Проф. М. Х. Хафизова. 22–23 окт. 2004 г. –Алматы, 2004. -С. 88–90.
- Ермоленко Н. А., Ермаков А. Ю., Бучнева И. А. Лечение эпилептических энцефалопатий с электрическим статусом медленного сна//Журнал «Медицинский совет».- 2008.- № 3–4.
- Зенков Л. Р., Притыко А. Г. Фармакорезистентные эпилепсии (рук. для врачей). -М.: «Медпресс информ», 2003. -197 с.
- Карлов В. А. Эпилептическая энцефалопатия. Журнал неврологии и психиатрии 2006; 2: 4–9.
- Киссин М. Я. Клиническая эпилептология. М.: ГЭОТАР-Медиа. 2009. 256 с.
- Кеппра в лечении эпилепсии: эффективность и переносимость/ К. Ю. Мухин, С. В. Пилия, В. А. Чадаев и др.// Журнал неврологии и психиатрии.- М., 2005.- № 1.- С.49–51.
- Лихтерман Б. Новое в диагностике и лечении эпилепсии. Фундаментальные науки — клинической эпилептологии// Медицинская газета.- М., 2005.- № 77 (5 окт.).- С.7.
- Моно и политерапия эпилепсии новым препаратом конвульсофином. // Медикал-экспресс. Центрально-азиатский медицинский журнал для практикующих врачей. -2002. -№ 1. (4). 473. — С. 46–47.
- Мухин К. Ю. Петрухин А. С. Миронов М. Б. Эпилептические синдромы. Диагностика и терапия. Справочное руководство для врачей. Москва 2008. с57–77.
- Петрухин А. С., Мухин К. Ю., Калинина Л. В. Ламиктал: поли- и монотерапия эпилепсии //Журнал Психиатрия и психофармакотерапия. -2004. -№ 1. -С. 3–7.
- Шамансуров Ш. Ш., Гулямова Д. Н. Нейропротективное действие глицина при судорожных состояниях у детей. В сб.: Актуальные проблемы неврологии //Международная конф., посв. 70-летию со дня рожд. проф. М. Х. Хафизова. 22–23 окт. 2004 г. -Алматы, 2004. -С. 306–307.
- Abend N. S., Dlugos D. J. Nonconvulsive status epilepticus in a pediatric intensive care unit.// Pediatr. Neurol. 2007 V. 37 p. 165–170.
- Andrade-Machado R, Benjumea-Cuartas V, Jaramillo-Jimenez E. Lacosamide in lennox-gastaut syndrome: case report // Clin Neuropharmacol. 2012 May;35(3):148–9.
- Aydin O. F., Senbil N., Gurer Y. K. Nonconvulsive status epilepticus on EEG in a case with subacute sclerosing panencephalitis.// J. Child. Neurol/ 2006. V. 21 p. 256–260.
- Chien YH, Lin MI, Weng WC, Du JC, Lee WT. Dextromethorphan in the treatment of early myoclonic encephalopathy evolving into migrating partial seizures in infancy // J Formos Med Assoc. 2012 May;111(5):290–4. Epub 2012 May 14.
- Cross JH. The ketogenic diet in the treatment of Lennox-Gastaut syndrome // Dev Med Child Neurol. 2012 May;54(5):394–5. doi: 10.1111/j.1469–8749.2012.04276.x.
- Cusmai R, Martinelli D, Moavero R, Dionisi Vici C, Vigevano F, Castana C, Elia M, Bernabei S, Bevivino E. Ketogenic diet in early myoclonic encephalopathy due to non ketotic hyperglycinemia // Eur J Paediatr Neurol. 2012 Jan 17.
- de Haan TR, Bijleveld YA, van der Lee JH, Groenendaal F, van den Broek MP, Rademaker CM, van Straaten HL, van Weissenbruch M. M. Pharmacokinetics and pharmacodynamics of medication in asphyxiated newborns during controlled hypothermia. The PharmaCool multicenter study // BMC Pediatr. 2012 May 22;12:45.
- Fejerman N, Caraballo R, Cersósimo R, Ferraro SM, Galicchio S, Amartino H. Sulthiame add-on therapy in children with focal epilepsies associated with encephalopathy related to electrical status epilepticus during slow sleep (ESES). // Epilepsia. 2012 Apr 17. doi: 10.1111/j.1528–1167.2012.03458.x.
- Grimminck B, de Jong H, Sluzewski M, Obihara CC. A child with convulsions of unknown origin: posterior reversible encephalopathy syndrome // Ned Tijdschr Geneeskd. 2012;156(17):A3920.
- Lee EH, Yum MS, Jeong MH, Lee KY, Ko TS. A case of malignant migrating partial seizures in infancy as a continuum of infantile epileptic encephalopathy // Brain Dev. 2011 Dec 23.
- Lerman-Sagie T, Ben Zeev B, Kramer U. Immunoglobulin treatment for severe childhood epilepsy. // Pediatr Neurol. 2012 Jun;46(6):375–81.
- Li Y, Jenny D, Castaldo J. Posterior reversible encephalopathy syndrome: clinicoradiological spectrum and therapeutic strategies // Hosp Pract (Minneap). 2012 Feb;40(1):202–13.
- Pawar PS, Noviawaty I, Zaidat OO. Unusual case of intra-arterial doxorubicin chemoembolization-associated posterior reversible encephalopathy syndrome // Neurologist. 2012 Jan;18(1):49–50.
- Romem A, Galante O, Shelef I, Almog Y. Posterior reversible encephalopathy syndrome complicating septic shock.// Isr Med Assoc J. 2011 Dec;13(12):776–8.
- Thammongkol S, Vears DF, Bicknell-Royle J, Nation J, Draffin K, Stewart KG, Scheffer IE, Mackay MT. Efficacy of the ketogenic diet: which epilepsies respond? // Epilepsia. 2012 Mar;53(3):e55–9. doi: 10.1111/j.1528–1167.2011.03394.x.
- Veeramah KR, O’Brien JE, Meisler MH, Cheng X, Dib-Hajj SD, Waxman SG, Talwar D, Girirajan S, Eichler EE, Restifo LL, Erickson RP, Hammer MF. De novo pathogenic SCN8A mutation identified by whole-genome sequencing of a family quartet affected by infantile epileptic encephalopathy and SUDEP // Am J Hum Genet. 2012 Mar 9;90(3):502–10. Epub 2012 Feb 23.
- Writzl K, Primec ZR, Stražišar BG, Osredkar D. Early onset West syndrome with severe hypomyelination and coloboma-like optic discs in a girl with SPTAN1 mutation // Epilepsia. 2012 Jun;53(6):e106–10.