From the history of the disease
Since the classic symptoms of chorea are erratic arm movements and an unsteady gait accompanied by dancing movements, it is not surprising that the Greek word chorea, which means “dance,” eventually became the name for this disease.
One of the first doctors to describe this serious illness was Paracelsus (1493-1541). Describing the phenomena of religious ecstasy in the form of the dance of St. Vitus, popular in the Middle Ages and the Renaissance, he discovered people demonstrating severe pathological symptoms. Much later, already in the 17th century, the doctor T. Sydenham described chorea, which appears in childhood and was named in his honor. And only in 1872, D. Huntington (Huntington) described a hereditary form of chorea that debuts in adults. Now this disease is called chorea or Huntington's disease (abbreviated as HD).
In 1993, an international team of genetic researchers discovered a mutation that causes HD.
Illness in childhood and adolescence
Chorea with sudden onset in childhood is most often caused by autoimmune and infectious etiologies. Slowly developing chorea syndrome in children and adolescents is mainly caused by cerebral palsy, less often by a neurodegenerative or metabolic disease, Sydenham's chorea after infection with group A β-hemolytic streptococcus, and even more rarely by benign hereditary chorea.
Causes of chorea
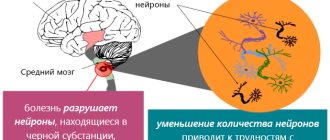
We are talking about serious genetic changes: this is the HNT gene, which is responsible for the development of pathology. Its location is the fourth chromosome. It has the ability to encode a special protein called huntingtin. How it works normally is unknown to science, but if it changes due to genetic abnormalities, HD occurs.
As the protein changes its structure, it causes nerve cells in the human brain to begin to function incorrectly and eventually die. Huntingtin is a complex substance, so the course of the disease is complex and multivariate. Modern researchers continue to actively study Huntington's chorea, trying to better understand the specifics of the basic mechanisms of its development.
The mutation that causes chorea is contained in all cells of the patient’s body, so we are talking about a classic hereditary disease.
Diagnostic Basics
Diagnosis is carried out using physical examination and psychological examination methods, which help to establish the main signs of the disease and assess the extent of the disease. *MRI image (on the left is a person suffering from Huntington’s chorea, on the right is a healthy person)
Of the instrumental diagnostic methods, the main place is occupied by computer and magnetic resonance imaging, which visualize the site of brain damage. Thus, the results of a CT or MRI show an area of atrophy or gliosis in the region of the striatum.
Using magnetic resonance spectroscopy, one more diagnostic sign can be determined - an increase in lactate levels in the tissue of the basal ganglia.
Of the screening methods, the “gold standard” is genetic testing, which is based on identifying an increased number of CAG trinucleotide residues in the HD gene. In the case when their number is 38 or more, the disease will occur over time in 100%. At the same time, there is a pattern that the greater the number of residues, the earlier chorea will appear.
Symptoms of chorea
The time of appearance of the first symptoms of chorea is middle age (period from 35 to 55 years). About 10% of clinical cases of HD occur in childhood and adolescence. In such situations, experts talk about juvenile chorea.
The development of the disease often occurs latently and is unnoticeable at first, so it can be diagnosed late.
The symptoms of chorea are varied. For example, it can manifest itself with a combination of symptoms (the patient’s mood constantly changes and difficulties in thinking arise). The development of symptoms depends on age and severity of the pathology. Some patients demonstrate predominantly motor disorders with mild mental disorders, while others, on the contrary, show anxiety, signs of depression or other mental disorders. The exact time when the first symptoms appear is difficult to determine.
Movement disorders
Movement disorders associated with chorea are usually called:
- actually, trochee;
- bradykinesia;
- dystonia.
These symptoms prevent the patient from being in a natural physiological position, disrupting the processes of walking and balance. “Dance” is expressed in involuntary movements that a person cannot control. Bradykinesia manifests itself in the slowness and complexity of voluntary movements, and dystonia manifests itself in the formation of unnatural and awkward poses, accompanied by “twisting” or twitching of the limbs. Also, movement disorders that occur with Huntington's chorea include dysfunction of the eye muscles, problems with swallowing and unclear speech.
Motor disturbances in the manifestations of chorea in adults are characterized by speed, pretentiousness and spontaneity. In this case, any muscle groups can be involved, and control of movements is absolutely impossible.
Usually, the onset of a pathological process is indicated by deviations observed during movements of the facial muscles. Patients grimace, stick out their tongues, raise their cheeks, curl their lips into a “tube,” frown and wink ridiculously. The progression of the pathology is accompanied by the appearance of involuntary movements in other muscles. Patients begin to quickly bend and straighten their fingers, then similar symptoms affect the lower extremities. Sometimes movements occur in the legs and arms at the same time.
Children suffering from chorea, on the contrary, demonstrate sluggish movements. They are very slow, and the child’s speech is slurred, with incorrect pronunciation of sounds and words, impaired speed and rhythm. From the motor sphere, oscillatory movements of the eyeballs are typical. The child cannot normally move his gaze from one object to another and focus his vision on a specific object.
When the disease begins in childhood and adolescence, experts note bradykinesia and muscle stiffness as the main symptoms. Children do not demonstrate violent movements, as with classical chorea in adults. Also, juvenile chorea is characterized by disturbances in behavior and learning with rapid progression of the entire symptom complex.
When the disease begins in a later age, chorea itself is its leading symptom, and its heredity is very easy to identify. Most often, the parents of an adult patient are no longer alive, since they simply do not live to see the moment when their offspring begin to experience severe symptoms of chorea.
Free consultation on training issues
Our consultants are always ready to tell you about all the details!
Clinical guidelines for Huntington's chorea
The list of basic recommendations should be compiled individually after examining the sick person by a psychiatrist and neurologist. The main clinical recommendations for Huntington's chorea are highlighted, which must be followed while in the hospital or when the patient is at home. The patient should regularly receive drugs that reduce the activity of dopamines in the cerebral cortex, the dose being increased every 3 days.
Parents, one of whom has been diagnosed with a genetic disease, are not recommended to have children. The probability of inheriting Huntington's chorea exceeds 50%; if pregnancy occurs, a DNA test is required for the unborn child. Statistically, people with Huntington's chorea most often die from pneumonia, cardiac activity, or other congestive processes in the body, so it is important to promptly treat any secondary diseases.
Mental changes
The psyche in HD undergoes gradual pathological changes. A decrease in a person’s ability to fully think is characterized, first of all, by problems with memory and a decrease in the critical threshold for perceiving one’s own state. The patient becomes anxious, irritable or, conversely, apathetic. In severe cases, hallucinatory or delusional syndromes develop. Suicidal behavior is also common in the context of severe depression.
Also, the following mental symptoms may appear:
- aggressive outbursts;
- insomnia;
- impulsiveness and spontaneity in behavior;
- social alienation.
Delusions and hallucinations are much less common. As for mental disorders, as the disease progresses, patients gradually lose memory acuity, logic and concentration. They find it difficult to make independent decisions and give clear answers to even the simplest questions. Over time, they learn new information worse and worse, which is an obvious sign of progressive dementia.
Stages of flow
Dr. A. Scholsen from Georgetown University (USA) proposes five stages of chorea development as a classification:
- early The patient has been diagnosed with Huntington's chorea, but so far his full functions have not been impaired;
- intermediate early. A person is able to work and have normal contact with society, but difficulties in thinking, movement and behavior are already making themselves felt. Patients can still cope with everyday activities and work;
- intermediate late. The patient is no longer able to work, but he still does household chores himself;
- late initial. The patient loses independence, but at home retains a number of functions - subject to the regular participation of other people in his daily life;
- late. Help is needed all the time, including professional care. Note that the late stage of the disease requires a special approach and skills from the people around the patient.
Huntington's disease
Symptoms and treatments
Huntington's Disease
(David Rubinsztein, Serious Science). Translation: PostScience.
Huntington's disease (also Huntington's disease) is a devastating autosomal dominant neurodegenerative disorder. The chances that a carrier will pass it on to one of their children are 50%.
Huntington's disease is one of the most common neurological diseases caused by a single gene mutation. The features of this disease - deposits of protein aggregates in nerve cells - are characteristic of Alzheimer's disease and Parkinson's disease.
Image of the disease process - a neuron affected by inclusion bodies // wikipedia.org
Causes of Huntington's disease
Huntington's disease is caused by a trinucleotin CAG repeat expansion in the gene encoding the huntingtin protein. Healthy people have fewer than 36 CAG repeats, the sequence looks like this: CCAGCAGCAGCAGCAGCAGCAGCAGCAGCAG... People with Huntington's disease have 36 or more of these repeats. When CAG repeats are translated into the amino acid glutamine, mutant huntingtin develops an abnormally long polyglutamine tract. This type of mutation is seen in eight other neurodegenerative diseases.
An extended polyglutamine tract imparts toxic properties to huntingtin. They may be due to the mutant protein's tendency to aggregate or because the mutant huntingtin interferes with the normal functioning of other proteins in the cell. This leads to neurodegeneration, especially noticeable in the caudate nucleus, putamen and cerebral cortex.
Symptoms of Huntington's disease: chorea
At the clinical level, the patient exhibits abnormal chaotic movements, decreased cognitive abilities (a form of dementia), and psychiatric abnormalities. The most obvious movement disorder seen in Huntington's disease is called chorea - abnormal short and irregular uncontrolled movements. Psychiatric symptoms of illness, such as depression, are partly related to the biology of the disease and are not always the patient's response to its presence.
Huntington's disease usually appears in midlife, around age 40. However, in cases with a very high number of repeats, the disease may appear in early childhood. In some cases, when the number of CAG repeats is close to 36, the disease manifests itself towards the end of life. The longer the chain of trinucleotide repeats, the earlier signs of the disease appear. Symptoms of the disease are similar in all patients, although there may be some differences at the initial stage. The disease continues for 15–20 years until the death of the patient.
History of Huntington's disease research
The disease is named after the American physician George Huntington, who described it in detail in 1872. “On Chorea” was the first of two articles by Huntington, in which he carefully described the symptoms of the disease that he observed in a family living on Long Island.
However, there are earlier descriptions of Huntington's disease. James Guzella first made the connection between the disease-causing gene and the short arm of the fourth human chromosome. This is the first classic example of how the location of a gene on a specific part of a chromosome can be discovered based on the study of families. Guzella and a large consortium's subsequent identification of the disease-causing mutation and gene enabled further precise genetic testing and provided a key resource for modeling the disease in cells and animals, which is critical for developing treatments.
Treatment of Huntington's disease
There is currently no known treatment to alleviate human neurodegeneration, but tetrabenazine may improve some movement disorders. Tetrabenazine is not thought to reduce the level of neurodegeneration in Huntington's disease. Chorea is caused by an excess of the neurotransmitter dopamine, tetrabenazine reduces its activity and reduces the symptom.
Numerous treatments are currently being developed to treat Huntington's disease at the mechanistic level. These include strategies to reduce mutant protein expression using antisense techniques (in clinical trials) and activation of autophagy. Antisense strategies involve nucleic acid oligonucleotides. They have sequences complementary to the Huntington's disease gene and reduce the amount of huntingtin synthesized. This strategy is quite rational, since the main driver of the disease is mutant huntingtin.
Prevalence of Huntington's disease
The disease affects 1 in 10,000 people in populations of European ancestry. Most often, Huntington's disease occurs in population isolates (in Venezuela), less often in some populations (for example, in the Japanese). Differences in the prevalence of the disease in populations are associated with the number of carriers of the gene in these groups. This is a consequence of historical events, including random increases or decreases in Huntington's disease carriers in population isolates.
The protective role of autophagy
In the laboratory, we have focused on the protective functions of autophagy in Huntington's disease and related neurodegenerative conditions. Autophagy is a process in which the internal components of a cell are delivered into its lysosomes or vacuoles and undergo degradation in them.
We found that intracellular aggregation-prone proteins (like mutant huntingtin) are substrates of autophagy. Importantly, we were the first to show that drugs that stimulate autophagy also stimulate the removal of toxic proteins. These are mutant huntingtin, mutant ataxin-3 (causing the most common spinocerebellar ataxia), alpha-synuclein (in Parkinson's disease), and wild-type and mutant tau proteins (associated with Alzheimer's disease and various types of frontotemporal dementia).
We have expanded our research from cellular systems to demonstrating the effectiveness of such drugs in disease models in fruit flies, zebrafish, and mice. This concept was subsequently confirmed by many research groups in various neurodegenerative diseases.
Our challenge is to develop this strategy into clinical reality. We have conducted a number of studies to identify new drugs that induce autophagy. My colleague Dr. Roger Barker and I have completed testing one of the identified drugs in patients with Huntington's disease.
Protein aggregate in the mouse brain (marked with arrows) // serious-science.org
Studying the functions of huntingtin and modern therapy
There are many ongoing research projects that are contributing to the study of the disease. First, the most active question being explored is how mutant huntingtin causes disease. To answer this, we need to use methods from structural biology, biophysics, genetic scanning, cell biology and animal models. Some groups are focusing on studying the disease at the biochemical level, trying to understand the structure of the mutant protein and its early aggregating species. Others are using cellular, neural, and stem cell models to understand what the mutant protein does. They are complemented by studies on animals: worms, fruit flies, zebrafish, mice, rats and even primates and sheep. This is necessary to develop models that will allow us to understand the disease at the organism level. Therapeutic strategies can be tested in such models.
Secondly, we need to understand what the functions of normal huntingtin are - they are poorly understood. To shed light on these functions, research groups are using different approaches based on cellular modeling. This could impact therapeutic strategies and/or our overall understanding of how the cell works.
The third goal is to identify potential therapeutic targets for disease relief and improve existing treatment strategies. Various research groups are working on this issue; they use chemical and genetic scanning techniques to identify new targets and drug candidates.
The fourth goal is to identify and characterize biomarkers of disease progression to facilitate clinical trials. This will make it possible to track the benefits of any therapeutic strategy. It would be useful to have a very sensitive scale of disease progression with a short time interval. This is important for those who are carriers of the gene for the disease, but do not yet have obvious signs and symptoms. In this case, it will be possible to test the effects of potential therapeutics that slow the progression of the disease.
About the author: David Rubinstein – Professor of Molecular Neurogenetics; Deputy Director, Cambridge Institute for Medical Research.
Portal “Eternal Youth” 04/21/2017
Forms of the disease
Based on the specifics of symptoms, the following forms of chorea are distinguished:
- hyperkinetic. It is characterized by spontaneous motor acts, which, as noted earlier, are not subject to conscious control. Also, speech disturbances due to hypertonicity of the facial muscles are typical for the hyperkinetic form of chorea. Sometimes patients have convulsions and uncontrolled oculomotor acts during sleep;
- akinetic-rigid. Severe muscle hypertonicity;
- mental. Dementia gradually develops, and the patient's personality undergoes destructive changes.
Psychotic phenomena for the latter form are also a characteristic feature.
Non-drug treatment
In some cases, patients respond well to non-drug therapy in the form of:
- psychotherapeutic influence;
- exercise therapy;
- classes with a qualified speech therapist;
- breathing exercises;
- occupational therapy.
Regular use of these methods (of course, if the patient’s condition allows) can significantly reduce the intensity of pathological symptoms both physically and mentally. It has been proven that patients’ psycho-emotional background improves, they begin to better control their voluntary movements. Walking becomes more stable, as do swallowing and balance.
Physical exercise is one of the effective ways to slow down the development of movement disorders. There are special exercise therapy programs developed for such patients, which have repeatedly shown their effectiveness.
Classification
Extrapyramidal hyperkinesis is:
- Primary (for various neurodegenerative diseases, for example, Huntington's disease , neuroacanthocytosis , spinocerebellar ataxia ).
- Secondary (a symptom of autoimmune, vascular and metabolic diseases, heavy metal intoxication, encephalitis of various etiologies).
According to their prevalence, choreic hyperkinesis is divided into:
- Focal.
- One-sided.
- Generalized.
Huntington's chorea, what is it? This disease is associated with the destruction of neurons in the subcortical nuclei and cortex. The disease manifests itself as a combination of movement disorders, dementia and progressive cognitive impairment. Since chorea is not the only manifestation of the disease, the term "Huntington's disease" is more correct than the term "chorea". The disease appears at the age of 40-50 and affects both sexes equally. The main manifestations of the disease are steadily progressing.
Photos of symptoms
Already at an early stage of Huntington's disease, in addition to motor disorders, a pronounced speech disorder (slow arrhythmic speech) appears; a swallowing disorder appears in the later stages and becomes the cause of aspiration , asphyxia and pneumonia . Changes in the mental sphere are represented by impaired memory, attention, phobic disorders and depression with suicidal attempts.
Among the secondary forms, minor chorea (synonymous with Sydenham's chorea), which is a complication of rheumatism, . This is a generally recognized variant of rheumatic lesions of the nervous system, which is why rheumatic chorea is also called neurorheumatism . The first descriptions of an epidemic of rheumatic chorea were made in 1418, and a complete description of the disease was given by Sydenham 200 years later. In 1831, Bright established a connection between minor chorea and rheumatic fever and described severe forms of pseudoparalytic minor chorea.
In recent years, thanks to antirheumatic treatment, Sydenham's chorea is very rare, and rheumatism itself is considered by rheumatologists as a disease that is losing its relevance. In the early 80s, neurorheumatism (St. Vitus' dance) was diagnosed in 36% of children with rheumatism, and currently in 16% of patients. In this case, hemichorea (hyperkinesis of one side) and muscle hypotonia (moderate and severe), impaired coordination, handwriting and speech, increased tendon reflexes and emotional lability predominate.
Drug treatment
Doctors use strong medication treatment in severe cases of chorea. One popular drug is tetrabenazine. It was developed specifically for the treatment of Huntington's chorea as a drug that reduces hyperkinesis.
Antipsychotics and drugs used for Parkinson's disease are also prescribed. The administration of antiparkinsonian drugs improves motor functions, and antipsychotics (for example, haloperidol, chlorpromazine, clozapine, etc.) alleviate the condition of patients with delusional and hallucinatory phenomena. If the patient is depressed, he is prescribed antidepressants; for insomnia, sedatives and sedatives are prescribed.
General information
Chorea, what kind of disease? Chorea (or choreic hyperkinesis) is continuous, fast, chaotic movements. Hyperkinesis involves the distal parts of the arms and legs, facial muscles, muscles of the larynx and trunk. These movements are violent and resemble grimacing, antics and antics. Hyperkinesis is associated with damage to brain structures united in the extrapyramidal system. These movement disorders significantly limit the patient's capabilities, leading to social isolation. Common forms of chorea are Huntington's chorea and rheumatic chorea (synonymous with St. Vitus' dance and Sydenham's chorea), which have different origins and prognoses.
If we consider Huntington's syndrome, Wikipedia gives the following definition of this disease: “... a genetic disease of the nervous system, which is characterized by progressive hyperkinesis and mental disorders.” The disease occurs with the destruction of brain neurons, which is manifested by a decrease in mental abilities and progressive movement disorders, and this invariably leads to impairment of the patient’s physical capabilities and disability. This is an incurable, progressive hereditary disease.
Rheumatic chorea or St. Vitus' dance associated with rheumatism has a favorable prognosis for treatment . Wikipedia gives the following definition: “... characterized by erratic and jerky movements that are similar to ordinary facial movements, but differ in amplitude and intensity, and therefore appear pretentious and grotesque, reminiscent of a dance.”
Also hyperkinesis associated with damage to the extrapyramidal system include torticollis, tremor, dystonia, athetosis, tics, myoclonus. All these conditions are characterized by hypotonic-hyperkinetic syndrome - that is, they combine hyperkinesis (involuntary, violent movements) and decreased muscle tone. The function of the extrapyramidal nervous system is to create conditions for movement (distributing muscle tone and preparing muscles for movement), ensuring posture and performing stereotypical reflex movements. All these functions are disrupted when the function of the extrapyramidal system is disrupted.