Причины мышечной дистрофии Эрба-Рота
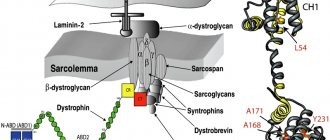
Причиной развития данной патологии является генетический дефект в гене 13q12, 17q12-q21.33, 4q12 и 5q33. Это обозначает, что в мышечной клетке образуется недостаточное количество ферментов. Именно они нужны клетки для последующего строительства белка. Выходит, что по причине недостаточности белка, повышается проницаемость мембран клеток. То есть, синтез саркогликанов нарушен, поэтому и нарушен белковый комплекс дистрофин-гликопротеин.
Белковый комплекс дистрофин-гликопротеин обеспечивает связь клеточного скелета сократительных элементов мышечных волокон миофибрилл с внеклеточными тканевыми структурами. В результате дефицита саркогликанов баланс аминокислот и ферментов в мышечных волокнах нарушается. Каркас мышечной клетки создает белок дистрофин. Каркас мышечной клетки похож на сетку Рабицу. Еще точнее на соты с медом. Если в одной соте меда не будет, то соседние соты увеличиваются в размере и немного сдвигаются одной стенкой в место пустой соты. Мед из увеличенных сот начинает вытекать.
Рис. 1. Генетический дефект при дистрофии Эрба-Рота
Точно так же начинает вытекать их мышечной клетки фермент креатинфосфокиназа. Он вытекает из клетки и попадает сначала в лимфу, затем из лимфы в кровь. Наличие в крови повышенного содержания креатинфосфокиназы показывает, что мышечная клетка в беде, т.е в ней нарушены биохимические реакции.
Для чего нужна креатинфосфокиназа мышечной клетки в достаточном количестве? Креатинфосфокиназа нужна мышечной клетки для того, чтобы митохондрия могла создавать энергию. Энергию в мышечной клетке создает митохондрия.
Мышечная слабость при мышечной дистрофии Эрба-Рота создает недостаточное количество креатинфосфокиназы внутри мышечной клетки.
Типы поясно-конечностной мышечной дистрофии
Ниже представлен список типов ПКМД.
Для типа 1 характерно наследование по доминантному признаку, т.е. требуется только одна мутация для проявления заболевания.
Для типа 2 характерно наследование по рецессивному признаку, т.е. требуется две мутации в гене – по одной от каждого родителя.
Некоторым типам ПКМД вместо чисел присвоены названия.
Типы ПКМД по отдельным названиям:
- миопатия Бетлема (мутация в гене collagen 6, доминантная)
- кальпаинопатия (мутация в гене calpain, рецессивная, другое название — LGMD2A)
- дисферлинопатия (мутация в гене dysferlin, рецессивная, другое название – LGMD2B)
- миофибриллярная миопатия (мутации в генах desmin, alpha-B crystallin, myotilin, ZASP, filamin C, BAG3 или SEPN1; все доминантные кроме desmin-типа, который может быть как доминантным, так и рецессивным)
- саркогликанопатии (мутация в гене sarcoglycan; рецессивная; другие названия — LGMD2C, LGMD2D, LGMD2E, LGMD2F)
- ZASP-связанные миопатии (мутация в гене ZASP; доминантная; является формой миофибриллярной миопатии)
Доминантные типы ПКМД по номерам:
- LGMD1A / ПКМД1А (мутация в гене myotilin)
- LGMD1B / ПКМД1B (мутация в гене lamin A/C)
- LGMD1C / ПКМД1С (мутация в гене caveolin)
- LGMD1D / ПКМД1D (мутация в гене DNAJB6)
- LGMD1E / ПКМД1E, также называемая десминовая миопатия – тип миофибриллярной миопатии (мутация в гене desmin)
- LGMD1F / ПКМД1F (мутация на 7-й хромосоме)
- LGMD1G / ПКМД1G (мутация на 4-й хромосоме)
- LGMD1H / ПКМД1H (мутация на 3-й хромосоме)
Рецессивные типы ПКМД по номерам:
- LGMD2A / ПКМД2A (мутация в гене calpain)
- LGMD2B / ПКМД2B (мутация в гене dysferlin)
- LGMD2C / ПКМД2C, также называется SCARMD1 (мутация в гене gamma sarcoglycan)
- LGMD2D / ПКМД2D, также называется SCARMD2 (мутация в гене alpha sarcoglycan)
- LGMD2E / ПКМД2E (мутация в гене beta sarcoglycan)
- LGMD2F / ПКМД2F (мутация в гене delta sarcoglycan)
- LGMD2G / ПКМД2G (мутация в гене telethonin)
- LGMD2H / ПКМД2H (мутация в гене TRIM32)
- LGMD2I / ПКМД2I (мутация в гене FKRP)
- LGMD2J / ПКМД2J (мутация в гене titin)
- LGMD2K / ПКМД2K (мутация в гене POMT1)
- LGMD2L / ПКМД2L (мутация в гене ANO5)
- LGMD2M / ПКМД2M (мутация в гене fukutin)
- LGMD2N / ПКМД2N (мутация в гене POMT2)
- LGMD2O / ПКМД2O (мутация в гене POMGnT1)
- LGMD2Q / ПКМД2Q (мутация в гене plectin)
Источник Muscular Dystrophy Association
Что делает мышечная клетка, чтобы остановить выход креатинфосфокиназы наружу?
Чтобы сохранить нужные вещества, клетка вынуждена закрывать эти дыры. А их можно закрыть только теми веществами, которые больше этих дыр. Мышечная клетка начинает задерживать внутри себя жировые компоненты, которые больше этих дыр.
Чтобы удержать жировые компоненты около этих дыр, нужно приложить силу. Чтобы приложить силу, нужна энергия. Энергию создают митохондрии. Поэтому, чтобы спасти жизнь клетки и свою жизнь, митохондрии перемещаются от двигательных белков актина и миозина к стенкам, полу и потолку клетки. Актин и миозин остаются без части энергии. Клетка, перепуганная тем, что могут образовываться другие дыры в каркасе, озабочена тем, что создает внутри себя дополнительные жировые включения (на всякий случай). Этих включений становиться так много, что жир начинает зажимать двигательные белки клетки до полного их обездвиживания.
Рис. 2. Состояние мышечной клетки при мышечной дистрофии Эрба-Рота
Пояснение к фото:
A — изменение размеров мышечных волокон и некротических миофибрилл (стрелки).
B — кластер базофильных регенерирующих миофибрилл (стрелки).
C — иммуногистохимическое окрашивание дистрофина демонстрирует заметную потерю нормальной окраски плазматической мембраны.
ЛФК
Обязательной составляющей терапии мышечной дистрофии Эрба-Рота является комплекс ЛФК. Основные цели, которые преследуют упражнения:
- развитие и поддержание мышечного аппарата;
- правильное расслабление;
- предупреждение контрактур, по причине которых пациент утрачивает способность передвигаться;
- правильное дыхание;
- предотвращение сколиоза и иных подобных деформаций.
В курсе терапии используются разные по уровню активности физические и дыхательные упражнения, массажи. При патологиях опорно-двигательного аппарата комплекс ЛФК всегда подбирается индивидуально специалистом. Исключение составляют незначительные нагрузки в бассейне. В таком случае упражнения сначала показывает инструктор, а после нескольких занятий пациент может уже самостоятельно их повторять.
Как действует мой метод для восстановления нормального движения мышц?
Применяя мой метод, Никонова Николая, при дистрофии Эрба-Рота: растяжения мышцы, фиксация ее в определенном положении и надавливая на нее определенным техническим приемом, происходит освобождение двигательных белков от пресса жировых включений.
В электронный микроскоп я вижу увеличение присутствия жировых клеток и концентрацию митохондрий у стенок клеток, что подтверждает мои логические рассуждения.
Рис. 3. Биопсия мышцы при диагнозе мышечная дистрофия Эрба-Рота
Биопсия мышцы показывает выраженный фиброз эндомизиального фибиса (A) и инфильтрацию лимфоцитов (B). (C) показаны базофильные регенерирующие волокна. (D) Некротический миофиб инфильтрирован отмеченными лимфоцитами и гистиоцитами
До тех пор, пока жировые включения, своей тяжестью, не остановили работу двигательных белков, движение мышц есть. Пусть слабые, пусть быстро утомляемые, но движения мышц есть.
Как только жировые включения зажимают двигательные белки, то образуется обездвиживание. Если обездвиживание произошло в диафрагме, то останавливается дыхание. Если обездвиживание произошло в сердце, то останавливается сердце.
Восстановить работу гена я не могу, но …, воздействуя на мышцы моим методом, получилось уменьшить количество жировых компонентов внутри мышечной клетки у девочки с прогрессирующей мышечной дистрофией Эрба-Рота и часть митохондрий вернулось на свои места.
Мышцы стали сокращаться и дали возможность Насте начать передвигаться.
Сейчас Настя находиться у себя дома, ухудшения ее состояния нет. Даже есть некоторое улучшения – Настя стала плавать в бассейне.
Доктор Никонов
Неудобство в восстановлении мышечной дистрофии Эрба-Рота в том, что нужно постоянно периодически воздействовать на мышцы моим методом, чтобы не было чрезмерно больших жировых отложений в мышечных клетках.
При сахарном диабете люди всю жизнь испытывают неудобства, принимая инсулин, но несмотря на это живут полноценной жизнью (заводят семью и детей).
Горжусь моими знаниями, опытом и навыками!
Результат моих знаний по восстановлению нормальной работы мышц при мышечной дистрофии Дюшенна:
Мышечная сила появилась у Эмине, Сергея, Якоба. Они и другие мои пациенты ходят, как обычные здоровые люди!
Диагностика
На обследование идут к неврологу или ревматологу. В ходе осмотра врач проверяет функционирование нервной системы, рефлексы, чувствительность кожи, телосложение пациента. Для определения состояния мышц проводят электронейромиографию. При подозрении Эрба-Рота миодистрофии выполняют биопсию мускульных волокон для цитоморфологии. Нужен общий и биохимический анализ крови, исследование концентрации креатинфосфокиназы, состава мочи.
Доктор также даст направление к генетику для выявления генных мутаций, провоцирующих миодистрофию Эрба-Рота.
Обязательно проводят дифференциальную диагностику с другими мышечными дистрофиями – клиническое течение Эрба-Рота и Дюшена иногда может протекать практически одинаково.
Для исключения возможных осложнений болезни делают флюорографию, электрокардиограмму, УЗИ мышц, сердца, органов брюшной полости и малого таза, рентгенографию грудной клетки.
Диагностические признаки Эрба-Рота миодистрофии:
- мышцах рук/ног отмечается гипотония (ослабление тонуса);
- уменьшается объем пораженной мускулатуры;
- ухудшаются или исчезают в конечностях сухожильные рефлексы;
- по нервным волокнам продолжают передаваться импульсы;
- нейроны не повреждаются;
- в мышцах выявляют разной толщины мускульные волокна, соединительную ткань, отмирание миоцитов;
- в мышечной ткани дефицит креатинфосфокиназы;
- возможно изменение электролитного состава крови;
- через время после дебюта в крови нормализуется показатель креатинфосфокиназы.
На осложнения миопатии указывает воспаление легких, дистрофия миокарда, истончение гладких мышц внутренних органов. На ЭКГ выявляют ухудшение проводимости, изменение ритмичности сердца.
Симптомы при дистрофии Эрба-Рота
Привожу ключевые симптомы при данном диагнозе, который начинает развиваться у детей и подростков:
- Происходит задержка в начале самостоятельно ходьбы ребенка.
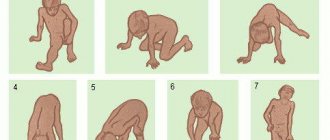
- Неудобная для больного походка, которая выглядит как ковыляние с ноги на ногу. Это еще называют как “утиный” тип ходьбы. Это происходит из-за симметричного ослабления мышц области беда.
- Ребенок часто спотыкается при передвижении и падает при беге, другими словам – дисбаланс и нестабильность.
- Сложности, возникающие при попытке встать с постели, стула. Также проявляются трудности при ходьбе на склонах, восхождениях и даже при спуске по лестнице.
- Наблюдается выпуклость лопаточных костей. Это происходит по причине ослабления передних зубчатых мышц грудной клетки больного и ромбовидных мышц спины.
- Уменьшается окружность талии. Это связано с тем, что при мышечной дистрофии Эрба-Рота происходит уменьшение тонуса поперечных мышц грудной клетки, живота и ileal-rib.
- Патологическая усталость у ребенка.
В результате прогрессирования болезни наблюдается постоянная общая слабость и ослабление мышечного корсета спины и мышц плечевого пояса. Эти процессы приводят к таким дефектам осанки как гиперлордоз. Пациентам с дистрофией становиться с каждым разом все труднее, как и удерживать предметы в руках, поднимать сами руки к верху. Что касается мимических лицевых мышц, то они тоже теряют свою подвижность. Это приводит к том, что наблюдается неполное закрытие век и выступ губ.
Процесс постепенного уменьшения мышечного тонуса приводит к неизбежному истончению и дряблости мышечной ткани больного на прогрессирующую дистрофию Эрба-Рота, заменяя ее жировой и фиброзной тканью, т.е. миодистрофией.
Симптомы заболевания на поздних стадиях
Серьезная потеря мышечной массы, сгибательная контура, сокращение сухожилий у больного и практически полная потеря глубоких сухожильных рефлексов нижних конечностей ребенка (колено и подошва).
Клинико-диагностические особенности мышечной дистрофии Дюшенна у детей
Актуальность. Среди миодистрофий наиболее часто в детском возрасте диагностируются прогрессирующую мышечную дистрофию Дюшенна, Эмери-Дрейфуса, Эрба-Рота [2, 8]. Миодистрофия Дюшенна — наследственное заболевание, занимает второе место в мире по частоте встречаемости (1:5000 новорожденных мальчиков) [1, 4]. Мышечная дистрофия Дюшенна является наиболее разрушительной из всех мышечных дистрофий [5].
Генетический фактор — несовершенный ген — передается ребенку от матери, но у них самих нарушения не развиваются. Имеется 50-процентный шанс, что у каждого сына женщины-носительницы генетического фактора разовьется болезнь и 50% шанс, что каждая ее дочь станет носителем этого фактора [3, 6].
Несмотря на почти вековую историю изучения данной патологии, вопросы их патогенеза, достоверной диагностики и лечения остаются до сего времени неразрешенными. Существует большое количество классификаций, но отсутствие точных данных о первичном биохимическом дефекте не дает возможности построить ее по рациональному принципу [1, 7].
Поражения внутренних органов значительно утяжеляет течение основного заболевания. Пациенты теряют способность самостоятельно передвигаться между 7-ю и 13-ю годами, а погибают, нередко, в подростковом возрасте или на III декаде жизни; при этом вовлеченными в патологический процесс оказываются практически все органы и системы организма [3].
Таким образом, анализ данных литературы убеждает в необходимости проведения тщательного соматического обследования пациентов с прогрессирующей мышечной дистрофией (ПМД) с целью как можно раньше выявить нарушения и принять меры для своевременной коррекции и поддержания функции жизненно важных органов.
В связи с вышеизложенным, целью данного исследования явилось изучение клинико-диагностических особенностей мышечной дистрофии Дюшенна у детей.
Материалы и методы исследования. Проведен клинический анализ 37 детей с диагнозом прогрессирующая мышечная дистрофия Дюшенна. Возраст детей варьировал от 3 до 15 лет, средний возраст составлял 7,8±0,48 лет. Возраст к началу заболевания в среднем составлял 4,3±0,36 лет и варьировал в пределах от рождения до 8 лет.
Биохимическое исследование включало определение уровня ферментов крови трансаминаз (АЛТ и АСТ), креатинфосфокиназы (КФК), лактатдегидразы (ЛДГ).
Генеалогическим методом обследовано 240 родственников I степени родства (родители, сибсы) детей с ПМД. Составлена подробная родословная, куда входили сведения о заболеваниях в 3 поколениях семьи. Генеалогический материал собирался по обеим родительским линиям путём перекрёстного опроса обоих родителей, иногда бабушек и дедушек.
Результаты и их обсуждение. При анализе акушерского анамнеза было установлено, что беременность протекала в 32,4% на фоне анемии, в 10,8% — токсикоза. 10,8% матерей во время беременности перенесли острые респираторные вирусные инфекции (ОРВИ). Обострение хронических заболеваний во время беременности регистрировалось у 2,7% матерей больных детей. Возраст матери при рождении ребенка с ПМД в среднем составил 26,0±0,85 лет.
Число членов семей, у которых отмечалась ПМД составило 13,5%.
По результатам наших исследований в 4 (10,8%) родословных выявлены случаи ПМД у родных братьев и в 1 случае (2,7%) у дяди по материнской линии.
Из анамнеза жизни установлено, что в 13,5% регистрируется родственный брак. Так же было отмечено, что в большинстве случаев дети были рождены от 2 и 3 беременности (2,2±0,19) или 2-3 родов (2,15±0,17).
Двое детей были из двойни, хочется обратить внимание, что один из детей двойни был здоров.
Роды в 89,2% случаев протекали нормально, в 5,4% случаев стремительно и по 1 (2,7%) случаю наблюдались длительный безводный период и оперативные роды. Вес при рождении детей составлял в среднем 3130±107,3 гр. В асфиксии были рождены 8 детей, что составило 21,6%. Обвитие пуповиной регистрировалось у 5,4% детей.
Анализ развития у детей психомоторных навыков показал, что дети в большинстве случаев поздно начинали удерживать голову (48,6%), сидеть (после 9 месяцев и позже 51,3%). Психомоторное развитие в 35,1% случаев не соответствовало возрастной норме ещё до дебюта проявлений миодистрофии Дюшенна.
В 78,4% случаях в предварительном диагнозе выставлялся ПМД. Из 37 наблюдаемых больных только 1 пациент находился на диспансерном учете, остальные же обращались впервые.
По данным литературы симптомы обычно проявляются в возрасте 2-5 лет, когда мышцы нижних частей тела и позвоночника поражаются первыми. Согласно нашим данным, первые симптомы заболевания проявились в среднем 4,6±0,35 лет.
Основными клиническими симптомами мышечной дистрофии Дюшенна являются: нежелание ходить или замедленная ходьба; ненормальная ходьба, часто походка вперевалку или качающаяся походка; ходьба на пальцах ног; неспособность нормально прыгать или бегать; трудности подъема по лестнице, при входе или выходе из автомобиля; частые падения. В связи с ходьбой на пальцах ног у этих детей развивается переднее наклонное положение таза и, соответственно, деформация спины.
У 67,6% обследованных детей наблюдались проблемы с зубами, характеризующиеся расширением челюсти и расширение промежутка между зубами.
При осмотре наблюдалось увеличение объема мышц, особенно икр; ноги, как правило, поражаются симметрично. Наиболее часто псевдогипертрофии наблюдались у детей старше 6 лет и по мере дальнейшего прогрессирования болезни имели тенденцию к уменьшению.
У 21,6% наблюдаемых детей развились ранние мышечные контрактуры и ретракция пяточных (ахилловых) сухожилий.
Мышечные атрофии первоначально локализовались в мышцах тазового пояса, с максимальной выраженностью в проксимальных отделах нижних конечностей. С возрастом имели тенденцию к распространению в восходящем направлении на мышцы плечевого пояса, спины и проксимального отдела верхних конечностей.
При детальном неврологическом осмотре в 97,3% случаев наблюдается утиная походка, активные движения ограничены, с затруднением самостоятельной ходьбы в 72,9% случаев.
Обращает на себя внимание снижение и утрата коленных рефлексов при длительном сохранении ахилловых рефлексов. У детей в более старшем возрасте наблюдается снижение рефлексов с m.biceps et m.triceps.
Во всех наблюдениях отмечается тотальная мышечная гипотония и снижение мышечной силы, более выраженные в ногах. Мышечная сила в руках составляла в среднем 2,2±0,07, в ногах 1,25±0,03.
Судорожный синдром отмечен у 2 детей, что составило 5,4%, тремор у 1 ребенка – 2,7%.
Отличительной чертой миодистрофии Дюшенна является сочетание атрофии мышц с патологией костно-суставной, сердечно-сосудистой и нейроэндокринной систем.
В соматическом статусе у обследованных детей с миодистрофией Дюшенна выражена костная патология на ранних стадиях заболевания типичными нарушениями являются: поясничный лордоз (100%), кифосколиоз (94,6%), сколиоз (5,4%), деформации грудной клетки по типу «килевидной» или «ладьевидной» груди (32,4%), высокий свод стопы (100%).
По мере прогрессирования процесса развивается эквиноварусная деформация стоп и контрактуры крупных суставов.
Сердечно-сосудистые расстройства клинически проявлялись лабильностью пульса, артериального давления (АД), глухостью тонов сердца. На электрокардиограмме (ЭКГ) регистрировались изменения миокарда, блокада ножек пучка Гиса.
Наиболее частыми нарушениями со стороны сердца среди обследованных больных являлись: выраженная тахикардия, аритмии и развитие сердечной недостаточности.
На ЭКГ у больных с миодистрофией Дюшенна регистрировался глубокий зубец Q в отведениях 2, 3, aVF, V6. и высокий зубец в отведении V6.
Полученные данные свидетельствуют о поражении миокарда в области задне-нижней и латеральной стенок левого желудочка.
У больных в развернутой стадии заболевания наиболее часто выявляется гипертрофическая кардиомиопатия (51,4%) и дилятационная (27%), реже встречались — пролапс митрального клапана и миксома левого желудочка (21,6%).
У 40,5% больных наблюдались нейроэндокринные нарушения, из них у 53,3% регистрировался синдром Иценко-Кушинга и у 46,7% адипозогенитальная дистрофия.
У 35,1% обследованных детей наблюдались психомоторные навыки не соответствующие возрасту, а в более старшем возрасте умственная отсталость.
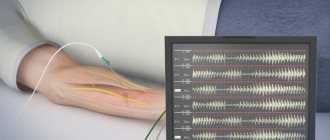
Среди 25 обследованных детей нами было проведено электронейромиографическое исследование (ЭНМГ) которое включало регистрацию биопотенциалов мышц кистей и стоп, измерение параметров прямого вызванного ответа мышцы (М — ответа) и поздно вызванных ответов.
Важным показателем состояния нервно-мышечного аппарата служит М-ответ. У всех обследованных детей данный показатель ниже как по tibialis anterior D, так и tibialis anterior S по сравнению с контрольной группы почти в 3,5 раза (Р<0,001). Скорость распространения возбуждения по нерву (СРВ) не отличалась от показателей контрольной группы с обеих сторон.
Таким образом, у всех обследованных детей в проксимальных отделах мышц нижних конечностей по данным ЭНМГ отмечалось первично-мышечное поражение с тенденцией к прогрессии и недостаточной реинервацией, которые были более выражение с возрастом.
Показатели лабораторных исследований были определены в сравнении с показателями здоровых детей (n=20). Данный анализ показал, что уровень КФК в основной группе был повышен в 71 раз, АЛТ в 15,2 раза, АСТ 8,4 раза, ЛДГ в 4,4 раза (рис. 1).
Высокая активность плазменных ферментов, прежде всего КФК отражает темп деградации миофибрилл в мышечных волокнах. Параллельно с повышением КФК наблюдалось увеличение концентрации и других ферментов цитолиза – лактатдегидразы и трансаминаз.
Увеличение уровня ЛДГ в крови свидетельствует о накоплении лактата крови, что в свою очередь приводит к гипоксии и вызывает чувство мышечной усталости, нарушает процесс тканевого дыхания.
Увеличение уровня трансаминаз (АЛТ и АСТ) в крови говорит всегда о большем, чем в норме, разрушении клеток соответствующего органа, в нашем случае мышцах.
Заключение. Таким образом, диагностика прогрессирующих мышечных дистрофий является сложной задачей, для решения которой необходимы создание и использование алгоритмов диагностики.
Диагноз прогрессирующая мышечная дистрофия Дюшенна основывается на данных клинико-генеалогического анамнеза, клинических особенностей заболевания (начало в 2-5 лет, симметричная атрофия проксимальных групп мышц, их развитие в восходящем направлении, псевдогипертрофия икроножных мышц, грубые соматические и нейроэндокринные нарушения, быстрое злокачественное течение заболевания).
Рис. 1. Лабораторные показатели у обследованных детей в сравнительном аспекте
Имеют значение также данные биохимических исследований (повышение в сыворотке крови уровня КФК в 71 раз выше от нормы на фоне повышения АЛТ, АСТ и ЛДГ), электромиографии и патоморфологии, при которых определяют первично-мышечный тип поражения.
Диагностика мышечной дистрофии Эрба-Рота
- Основывается диагностика рассматриваемого заболевания на физическом обследовании больных, исследовании семейной истории пациента и последующем анализе собранных данных.
- Проводится генетическое тестирование. Нужно для последующего точно определения мышечной дистрофии.
- Электронейромиография.
- Проводится биопсия мышечной ткани с биохимическим исследованием.
- Сдается общий анализ крови.
- Сдается анализ крови на креатинфосфокиназа.
- Анализ мочи больного.
Что касается электромиографии, то она позволяет исследовать не только степень нервно-мышечной передачи, но и определить уровень непосредственной мышечной возбудимости, что крайне важно для дифференциальной диагностики заболевания с патологиями нейропатических мышц.
Что такое поясно-конечностная мышечная дистрофия?
Примечание переводчика. В разных источниках название варьируется: поясно-конечностная МД и конечностно-поясная МД.
Поясно-конечностная мышечная дистрофия (ПКМД) – это не одно общее заболевание. Это целая группа заболеваний, поражающих мышцы, в основном расположенные в районе бёдер и плеч.
Плечевой пояс – это костная структура, которая окружает плечевую область.
(Информация из Википедии: плечевой пояс (пояс верхних конечностей) — совокупность костей (пары лопаток и ключиц) и мышц, обеспечивающих опору и движение верхних (передних) конечностей.)
Тазовый пояс – это костная структура, окружающая район бёдер.
Совокупно они называются «пояса конечностей». При ПКМД больше всего повреждаются мышцы, соединённые с костями этих поясов.
Термин «проксимальный» также используется для описания повреждённых при ПКМД мышц. Проксимальные мышцы – это мышцы, расположенные близко к центру тела. Дистальные мышцы – это более удалённые от центра тела мышцы (например, мышцы кистей или ступней). Дистальные мышцы при ПКМД поражаются на поздних этапах, но могут и сохранить свою функцию.
По данным на конец 2012 года насчитывается более 20 различных подтипов ПКМД. Это сложная и постоянно развивающаяся область исследований.
Лечение дистрофии Эрба-Рота
Следует сразу же отметить, что поврежденный ген я не могу восстановить, но…
Доктор Никонов
Воздействие по моему методу направлено на снижение интенсивности симптомов, замедление прогрессирования заболевания, повышение силы в мышцах, восстановление правильного движения во всех группах мышц.
Я не занимаюсь лечением мышечной дистрофии Эрба-Рота. Я занимаюсь восстановлением нормальной работы мышц. Поэтому лечебные процедуры, применяемые в стационарах и в других реабилитационных центрах, описывать в моей статье не буду. Скорее всего, вы их испробовали.
Моя статья носит ознакомительный характер. Информацию я взял из своих наблюдений и десятилетних изучений-открытий ученых со всего мира.
Медицинская справка
Заболевание представляет собой полиморфный вариант наследственной миодистрофии. От других разновидностей патологии оно отличается клинической картиной, течением и временем начала. Впервые описание недуга представил немецкий невролог В. Эрб в 1882 году. Одновременно с ним этой проблемой в России занимался В. Рот, которую позднее он обозначил как «мышечная сухотка». Именно по фамилиям двух ученых и было названо заболевание. В современной неврологии используется несколько его наименований — прогрессирующая мышечная дистрофия Эрба-Рота, конечностно-поясная мышечная дистрофия.
Патология начинает свое развитие, как правило, в детстве. Однако возраст появления первых симптомов может колебаться в диапазоне от 10 от 30 лет. Мужчины и женщины в равной степени страдают от проявлений мышечной дистрофии. Неврологи отмечают, что начавшийся в детстве недуг быстро прогрессирует, если сравнивать его течение в подростковом и взрослом возрасте. Кроме того, во втором случае он протекает в легкой форме.
Лечение
Болезнь неизлечима, поскольку только изучаются способы устранения генной аномалии. Терапевтические методы направлены на торможение прогресса миодистрофии Эрба-Рота, купирование симптомов, улучшения метаболизма. Упор делают на рациональное питание, прием витаминно-минеральных комплексов, массаж, ЛФК, физиотерапию.
Что назначают при Эрба-Рота миодистрофии:
- витамины E, группу B, D, C, A;
- аденозинтрифосфорную кислоту;
- внутримышечные инъекции экстракта алоэ;
- метаболики — Актовегин, Рибоксин, Тиоктовая кислота;
- препараты галантамина;
- антиоксиданты, антигипоксанты — средства триметазидина;
- введение стволовых фетальных клеток;
- хлорид кальция — для ионофореза;
- Прозерин — для электрофореза;
- антидепрессанты 4 поколения с эсциталопрамом, флуоксетином.
Справка! При болезни Эрба-Рота прекратили использовать «Ретаболил» и другие гормональные препараты нандролона. В ходе клинических наблюдений выявлено ускорение распада мускульных волокон.
При Эрба-Рота миодистрофии пациент должен ежедневно употреблять белковую пищу (мясо, рыбу) и свежие фрукты, овощи, питательные смеси с аминокислотами, L-карнитином. Их вещества используются организмом для построения мускульных волокон. Важно пить минеральную воду (разновидность и суточную норму врачи подбирают персонально). Разрешено после консультации с доктором применять народные средства.
Для улучшения состояния здоровья полезны:
- чай лапачо — кора муравьиного дерева;
- настой плодов шиповника;
- отвар девясила;
- мед;
- цветочная пыльца.
При Эрба-Рота миодистрофии делают ежедневно упражнения ЛФК, дыхательную гимнастику и массаж. Плавают не реже 1 раза/неделю. Методы используют для укрепления и увеличения объема мышц, нормализации питания клеток, микроциркуляции. Это улучшает метаболизм, сохраняет способность двигаться, убирает застой жидкости в органах, мягких тканях.
Принципы ЛФК при Эрба-Рота миодистрофии:
- нагрузку и упражнения подбирает врач согласно состоянию здоровья;
- темп выполнения и интенсивность умеренные или средние;
- тренируют все группы мышц;
- людям с ограничениями упражнения помогают делать близкие;
- длительность тренировки не должна превышать 20 минут;
- большой комплекс упражнений разбивают на 2―3 раза/день.
Поддерживающий массаж выполняют с легким нажимом минимум полчаса. Используют разминающие, поглаживающие, растирающие, точечные, продольные движения. При уходе за инвалидом надо чаще, интенсивнее массировать зону грудной клетки, чтобы предупредить развитие застойной пневмонии.
Для растяжения укороченных мышц делают элонгирующий массаж. При Эрба-Рота миодистрофии такой сеанс проводят от 20 минут, нажимают сильнее, используют растягивающие, разминающие, вибрирующие движения.
Осложнения лечат этиологическими лекарствами и методами в зависимости от того, какой орган затронут. Например, в случае пневмонии используют антибиотики, при дыхательной недостаточности назначают искусственную вентиляцию легких.
Профилактика
К первичной профилактике относят отказ от брака с кровным родственником, соблюдение правил безопасности при работе с вредными веществами. Людям с подтвержденным гетерозиготным носительством следует помнить о риске рождения детей с миодистрофией. Их ребенка врачи должны наблюдать с младенчества, а родителям надо ежедневно делать малышу гимнастику и массаж.
Важно! ЛФК, лечебное питание, массаж и методы физиотерапии помогают затормозить прогресс миодистрофии Эрба-Рота в любом возрасте. Проводится только профилактика осложнений.