Causes of Erb-Roth muscular dystrophy
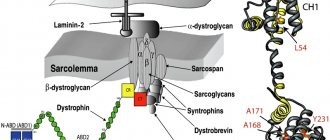
The cause of the development of this pathology is a genetic defect in the gene 13q12, 17q12-q21.33, 4q12 and 5q33. This means that an insufficient amount of enzymes is formed in the muscle cell. These are exactly what cells need for subsequent protein construction. It turns out that due to protein deficiency, the permeability of cell membranes increases. That is, the synthesis of sarcoglycans is impaired, and therefore the dystrophin-glycoprotein protein complex is impaired.
The dystrophin-glycoprotein protein complex ensures the connection of the cellular skeleton of the contractile elements of muscle fibers of myofibrils with extracellular tissue structures. As a result of sarcoglycan deficiency, the balance of amino acids and enzymes in muscle fibers is disrupted. The muscle cell framework creates the protein dystrophin. The framework of a muscle cell is similar to a chain-link mesh. Even more precisely, on honeycombs with honey. If there is no honey in one comb, then the neighboring combs increase in size and move slightly with one wall in place of the empty comb. Honey begins to flow out of the enlarged honeycombs.
Rice. 1. Genetic defect in Erb-Roth dystrophy
In the same way, the enzyme creatine phosphokinase begins to leak out of their muscle cells. It flows out of the cell and enters first the lymph, then from the lymph into the blood. The presence of increased levels of creatine phosphokinase in the blood indicates that the muscle cell is in trouble, that is, its biochemical reactions are disturbed.
Why is creatine phosphokinase needed in muscle cells in sufficient quantities? Creatine phosphokinase is needed by the muscle cell so that the mitochondria can create energy. Energy in a muscle cell is created by mitochondria.
Muscle weakness in Erb-Roth muscular dystrophy creates insufficient amounts of creatine phosphokinase within the muscle cell.
Types of limb-girdle muscular dystrophy
Below is a list of types of PCMD.
Type 1 dominant inheritance , i.e. only one mutation is required to manifest the disease.
Type 2 recessive inheritance , i.e. two mutations in the gene are required, one from each parent.
Some types of PCMD are given names instead of numbers.
Types of PCMD by individual names:
- Bethlem myopathy (mutation in the collagen 6 gene, dominant)
- calpainopathy (mutation in the calpain gene, recessive, another name is LGMD2A)
- dysferlinopathy (mutation in the dysferlin gene, recessive, another name is LGMD2B)
- myofibrillar myopathy (mutations in the desmin, alpha-B crystallin, myotilin, ZASP, filamin C, BAG3 or SEPN1 genes; all dominant except the desmin type, which can be either dominant or recessive)
- sarcoglycanopathy (mutation in the sarcoglycan gene; recessive; other names - LGMD2C, LGMD2D, LGMD2E, LGMD2F)
- ZASP-related myopathies (mutation in the ZASP gene; dominant; is a form of myofibrillar myopathy)
Dominant types of PCMD by numbers:
- LGMD1A / PKMD1A (mutation in the myotilin gene)
- LGMD1B / PKMD1B (mutation in the lamin A/C gene)
- LGMD1C / PKMD1C (mutation in the caveolin gene)
- LGMD1D / PKMD1D (mutation in the DNAJB6 gene)
- LGMD1E / PCMD1E , also called desmin myopathy - a type of myofibrillar myopathy (mutation in the desmin gene)
- LGMD1F / PKMD1F (mutation on chromosome 7)
- LGMD1G / PKMD1G (mutation on chromosome 4)
- LGMD1H / PKMD1H (mutation on chromosome 3)
Recessive types of PCMD by numbers:
- LGMD2A / PKMD2A (mutation in the calpain gene)
- LGMD2B / PCMD2B (mutation in the dysferlin gene)
- LGMD2C/PCMD2C , also called SCARMD1 (gamma sarcoglycan gene mutation)
- LGMD2D/PCMD2D , also called SCARMD2 (alpha sarcoglycan gene mutation)
- LGMD2E / PKMD2E (mutation in the beta sarcoglycan gene)
- LGMD2F / PKMD2F (mutation in the delta sarcoglycan gene)
- LGMD2G / PKMD2G (mutation in the telethonin gene)
- LGMD2H / PKMD2H (mutation in the TRIM32 gene)
- LGMD2I / PKMD2I (mutation in the FKRP gene)
- LGMD2J / PKMD2J (mutation in the titin gene)
- LGMD2K / PKMD2K (mutation in the POMT1 gene)
- LGMD2L / PKMD2L (mutation in the ANO5 gene)
- LGMD2M / PKMD2M (mutation in the fukutin gene)
- LGMD2N / PKMD2N (mutation in the POMT2 gene)
- LGMD2O / PKMD2O (mutation in the POMGnT1 gene)
- LGMD2Q / PKMD2Q (mutation in the plectin gene)
Source: Muscular Dystrophy Association
What does a muscle cell do to stop the release of creatine phosphokinase?
To preserve the necessary substances, the cell is forced to close these holes. And they can only be closed with substances that are larger than these holes. The muscle cell begins to retain fat components inside itself that are larger than these holes.
To keep the fatty components near these holes, you need to apply force. To exert force requires energy. Energy is created by mitochondria. Therefore, to save the life of the cell and their lives, mitochondria move from the motor proteins actin and myosin to the walls, floor and ceiling of the cell. Actin and myosin are left without some of the energy. The cell, frightened that other holes may form in the frame, is concerned about creating additional fatty inclusions inside itself (just in case). There are so many of these inclusions that the fat begins to squeeze the motor proteins of the cell until they are completely immobilized.
Rice. 2. State of the muscle cell in Erb-Roth muscular dystrophy
Explanation for the photo:
A - change in the size of muscle fibers and necrotic myofibrils (arrows).
B — cluster of basophilic regenerating myofibrils (arrows).
C , Immunohistochemical staining for dystrophin demonstrates marked loss of normal plasma membrane staining.
Exercise therapy
A mandatory component of the treatment of Erb-Roth muscular dystrophy is a complex of exercise therapy. The main goals of the exercises:
- development and maintenance of the muscular system;
- proper relaxation;
- prevention of contractures due to which the patient loses the ability to move;
- correct breathing;
- prevention of scoliosis and other similar deformities.
The course of therapy uses physical and breathing exercises and massages of varying levels of activity. For pathologies of the musculoskeletal system, the exercise therapy complex is always selected individually by a specialist. The exception is minor loads in the pool. In this case, the exercises are first shown by the instructor, and after several lessons the patient can repeat them independently.
How does my method work to restore normal muscle movement?
Using my method, Nikolay Nikonov, for Erb-Roth dystrophy: stretching the muscle, fixing it in a certain position and pressing on it with a certain technical technique, the motor proteins are released from the pressure of fatty inclusions.
Using an electron microscope, I see an increase in the presence of fat cells and a concentration of mitochondria near the cell walls, which confirms my logical reasoning.
Rice. 3. Muscle biopsy for the diagnosis of Erb-Roth muscular dystrophy
Muscle biopsy shows severe fibrosis of the endomysial fibis ( A ) and lymphocyte infiltration ( B ). ( C ) Basophilic regenerating fibers are shown. ( D ) Necrotic myofib infiltrated with marked lymphocytes and histiocytes
Until the fatty inclusions, with their heaviness, stop the work of motor proteins, there is muscle movement. They may be weak, they may tire quickly, but there are muscle movements.
As soon as fatty inclusions clamp motor proteins, immobilization occurs. If immobilization occurs in the diaphragm, breathing stops. If immobilization occurs in the heart, then the heart stops.
I cannot restore the functioning of the gene, but... by influencing the muscles with my method, it was possible to reduce the amount of fatty components inside the muscle cell in a girl with progressive Erb-Roth muscular dystrophy and some of the mitochondria returned to their places.
The muscles began to contract and made it possible for Nastya to begin to move.
Now Nastya is at home, her condition has not worsened. There is even some improvement - Nastya started swimming in the pool.
Doctor Nikonov
The disadvantage of restoring Erb-Roth muscular dystrophy is that you need to constantly periodically influence the muscles with my method so that there are no excessively large fat deposits in the muscle cells.
With diabetes, people experience inconvenience all their lives when taking insulin, but despite this they live a full life (have a family and children).
I am proud of my knowledge, experience and skills!
The result of my knowledge on restoring normal muscle function in Duchenne muscular dystrophy:
Emine, Sergei, and Jacob gained muscle strength. They and my other patients walk like ordinary healthy people!
Diagnostics
They go to a neurologist or rheumatologist for examination. During the examination, the doctor checks the functioning of the nervous system, reflexes, skin sensitivity, and the patient’s physique. To determine the condition of the muscles, electroneuromyography is performed. If Erb-Roth muscular dystrophy is suspected, a muscle fiber biopsy is performed for cytomorphology. A general and biochemical blood test, a study of the concentration of creatine phosphokinase, and the composition of urine are needed.
The doctor will also give a referral to a geneticist to identify gene mutations that cause Erb-Roth muscular dystrophy.
It is imperative to carry out differential diagnosis with other muscular dystrophies - the clinical course of Erb-Roth and Duchenne can sometimes proceed almost identically.
To exclude possible complications of the disease, fluorography, electrocardiogram, ultrasound of muscles, heart, abdominal and pelvic organs, and chest x-ray are done.
Diagnostic signs of Erb-Roth muscular dystrophy:
- hypotension (weakening of tone) is noted in the muscles of the arms/legs;
- the volume of the affected muscles decreases;
- tendon reflexes worsen or disappear in the limbs;
- impulses continue to be transmitted along the nerve fibers;
- neurons are not damaged;
- in the muscles, muscle fibers of different thicknesses, connective tissue, and the death of myocytes are detected;
- deficiency of creatine phosphokinase in muscle tissue;
- possible changes in the electrolyte composition of the blood;
- some time after the onset, the level of creatine phosphokinase in the blood normalizes.
Complications of myopathy are indicated by pneumonia, myocardial dystrophy, and thinning of the smooth muscles of internal organs. An ECG reveals deterioration in conduction and changes in heart rhythm.
Symptoms of Erb-Roth dystrophy
Here are the key symptoms for this diagnosis, which begins to develop in children and adolescents:
- There is a delay in the child starting to walk independently.
- An uncomfortable gait for the patient, which looks like hobbling from foot to foot. This is also called the “duck” type of walking. This occurs due to a symmetrical weakening of the muscles in the area of pain.
- The child often stumbles when moving and falls when running, in other words – imbalance and instability.
- Difficulties that arise when trying to get out of bed or chair. Difficulties also occur when walking on slopes, climbing, and even going down stairs.
- There is a convexity of the scapular bones. This occurs due to weakening of the patient’s serratus anterior muscles and the rhomboid muscles of the back.
- Waist circumference decreases. This is due to the fact that with Erb-Roth muscular dystrophy, there is a decrease in the tone of the transverse muscles of the chest, abdomen and ileal rib.
- Pathological fatigue in a child.
As the disease progresses, there is constant general weakness and weakening of the muscular corset of the back and shoulder girdle muscles. These processes lead to such postural defects as hyperlordosis. For patients with dystrophy, it becomes more and more difficult each time, as well as holding objects in their hands and raising their hands to the top. As for the facial muscles, they also lose their mobility. This results in incomplete closure of the eyelids and protrusion of the lips.
The process of gradual decrease in muscle tone leads to inevitable thinning and flabbiness of the patient’s muscle tissue with progressive Erb-Roth dystrophy, replacing it with adipose and fibrous tissue, i.e. myodystrophy.
Symptoms of the disease in later stages
Severe loss of muscle mass, flexion contour, contraction of the patient's tendons and almost complete loss of deep tendon reflexes in the child's lower extremities (knee and sole).
Clinical and diagnostic features of Duchenne muscular dystrophy in children
Relevance. Among the muscular dystrophies, progressive muscular dystrophy of Duchenne, Emery-Dreyfus, and Erb-Roth is most often diagnosed in childhood [2, 8]. Duchenne muscular dystrophy is a hereditary disease that ranks second in the world in terms of incidence (1:5000 newborn boys) [1, 4]. Duchenne muscular dystrophy is the most destructive of all muscular dystrophies [5].
The genetic factor - an imperfect gene - is passed on to the child from the mother, but they themselves do not develop disorders. There is a 50% chance that each son of a woman who is a carrier of a genetic factor will develop the disease, and a 50% chance that each of her daughters will become a carrier of this factor [3, 6].
Despite the almost century-long history of studying this pathology, questions of its pathogenesis, reliable diagnosis and treatment remain unresolved to this day. There are a large number of classifications, but the lack of accurate data on the primary biochemical defect does not make it possible to build it on a rational principle [1, 7].
Damage to internal organs significantly aggravates the course of the underlying disease. Patients lose the ability to move independently between the ages of 7 and 13, and often die in adolescence or in the third decade of life; in this case, almost all organs and systems of the body are involved in the pathological process [3].
Thus, an analysis of literature data convinces of the need to conduct a thorough somatic examination of patients with progressive muscular dystrophy (PMD) in order to identify disorders as early as possible and take measures for timely correction and maintenance of the function of vital organs.
In connection with the above, the purpose of this study was to study the clinical and diagnostic features of Duchenne muscular dystrophy in children.
Materials and methods of research. A clinical analysis of 37 children diagnosed with progressive Duchenne muscular dystrophy was carried out. The age of the children ranged from 3 to 15 years, the average age was 7.8±0.48 years. The age at the onset of the disease averaged 4.3±0.36 years and ranged from birth to 8 years.
The biochemical study included determination of the level of blood enzymes transaminases (ALT and AST), creatine phosphokinase (CPK), and lactate dehydrase (LDH).
Using the genealogical method, 240 first degree relatives (parents, siblings) of children with PMD were examined. A detailed pedigree was compiled, which included information about diseases in 3 generations of the family. Genealogical material was collected on both parental lines by cross-questioning both parents, and sometimes grandparents.
Results and its discussion. When analyzing the obstetric history, it was found that pregnancy occurred in 32.4% against the background of anemia, in 10.8% - toxicosis. 10.8% of mothers suffered acute respiratory viral infections (ARVI) during pregnancy. Exacerbation of chronic diseases during pregnancy was recorded in 2.7% of mothers of sick children. The average age of the mother at the birth of a child with PMD was 26.0±0.85 years.
The number of family members who had PMD was 13.5%.
According to the results of our studies, in 4 (10.8%) pedigrees, cases of PMD were identified in siblings and in 1 case (2.7%) in a maternal uncle.
From the life history it was established that in 13.5% a consanguineous marriage was registered. It was also noted that in most cases, children were born from the 2nd and 3rd pregnancies (2.2±0.19) or 2-3 births (2.15±0.17).
Two of the children were twins; I would like to point out that one of the twin children was healthy.
Childbirth in 89.2% of cases proceeded normally, in 5.4% of cases it was rapid and in 1 (2.7%) case a long anhydrous period and operative delivery were observed. The birth weight of children averaged 3130±107.3 g. 8 children were born due to asphyxia, which amounted to 21.6%. Umbilical cord entanglement was recorded in 5.4% of children.
An analysis of the development of psychomotor skills in children showed that children in most cases began to hold their heads late (48.6%) and sit (after 9 months and later 51.3%). Psychomotor development in 35.1% of cases did not correspond to the age norm even before the onset of manifestations of Duchenne muscular dystrophy.
In 78.4% of cases, the preliminary diagnosis was PMD. Of the 37 observed patients, only 1 patient was registered at the dispensary, while the rest came for the first time.
According to the literature, symptoms usually appear between the ages of 2 and 5 years, when the muscles of the lower body and spine are first affected. According to our data, the first symptoms of the disease appeared on average 4.6±0.35 years.
The main clinical symptoms of Duchenne muscular dystrophy are: reluctance to walk or slow walking; abnormal walking, often with a waddle or swaying gait; walking on your toes; inability to jump or run normally; difficulty climbing stairs, entering or exiting a car; frequent falls. Due to walking on their toes, these children develop anterior pelvic tilt and, accordingly, back deformity.
67.6% of the examined children had dental problems, characterized by widening of the jaw and widening of the gap between the teeth.
On examination, there was an increase in muscle volume, especially in the calves; the legs are usually affected symmetrically. Pseudohypertrophy was most often observed in children over 6 years of age and tended to decrease as the disease progressed.
21.6% of observed children developed early muscle contractures and retraction of the heel (Achilles) tendons.
Muscle atrophy was initially localized in the muscles of the pelvic girdle, with maximum severity in the proximal parts of the lower extremities. With age, they tended to spread in an upward direction to the muscles of the shoulder girdle, back and proximal upper limbs.
During a detailed neurological examination, a duck's gait is observed in 97.3% of cases, active movements are limited, with difficulty walking independently in 72.9% of cases.
Noteworthy is the decrease and loss of knee reflexes with long-term preservation of Achilles reflexes. In older children, there is a decrease in reflexes with m.biceps and m.triceps.
In all observations, total muscle hypotonia and a decrease in muscle strength, more pronounced in the legs, are noted. Muscle strength in the arms averaged 2.2±0.07, in the legs 1.25±0.03.
Convulsive syndrome was noted in 2 children, which amounted to 5.4%, tremor in 1 child – 2.7%.
A distinctive feature of Duchenne muscular dystrophy is the combination of muscle atrophy with pathology of the osteoarticular, cardiovascular and neuroendocrine systems.
In the somatic status of the examined children with Duchenne muscular dystrophy, bone pathology is expressed in the early stages of the disease; typical disorders are: lumbar lordosis (100%), kyphoscoliosis (94.6%), scoliosis (5.4%), “keeled” chest deformities "or "scaphoid" chest (32.4%), high arch (100%).
As the process progresses, equinovarus deformity of the feet and contractures of large joints develop.
Cardiovascular disorders were clinically manifested by lability of pulse, blood pressure (BP), and dullness of heart sounds. The electrocardiogram (ECG) recorded myocardial changes and bundle branch block.
The most common cardiac disorders among the examined patients were: severe tachycardia, arrhythmias and the development of heart failure.
On the ECG in patients with Duchenne muscular dystrophy, a deep Q wave was recorded in leads 2, 3, aVF, V6. and a high wave in lead V6.
The data obtained indicate myocardial damage in the region of the posteroinferior and lateral walls of the left ventricle.
In patients in the advanced stage of the disease, hypertrophic cardiomyopathy (51.4%) and dilated (27%) are most often detected; mitral valve prolapse and left ventricular myxoma (21.6%) are less common.
Neuroendocrine disorders were observed in 40.5% of patients, of which 53.3% had Itsenko-Cushing syndrome and 46.7% had adiposogenital dystrophy.
35.1% of the examined children had psychomotor skills that were not appropriate for their age, and at an older age, mental retardation.
Among the 25 children examined, we conducted an electroneuromyographic study (ENMG), which included recording the biopotentials of the muscles of the hands and feet, measuring the parameters of the direct evoked response of the muscle (M - response) and late evoked responses.
An important indicator of the state of the neuromuscular system is the M-response. In all examined children, this indicator was lower in both tibialis anterior D and tibialis anterior S compared to the control group by almost 3.5 times (P<0.001). The speed of propagation of excitation along the nerve (SPT) did not differ from the control group on both sides.
Thus, in all the examined children, according to ENMG, in the proximal parts of the muscles of the lower extremities, primary muscle damage was noted with a tendency to progression and insufficient reinnervation, which became more pronounced with age.
Indicators of laboratory tests were determined in comparison with indicators of healthy children (n=20). This analysis showed that the level of CPK in the main group was increased by 71 times, ALT by 15.2 times, AST by 8.4 times, and LDH by 4.4 times (Fig. 1).
High activity of plasma enzymes, primarily CPK, reflects the rate of degradation of myofibrils in muscle fibers. In parallel with the increase in CPK, there was an increase in the concentration of other cytolytic enzymes - lactate dehydrase and transaminases.
An increase in the level of LDH in the blood indicates the accumulation of blood lactate, which in turn leads to hypoxia and causes a feeling of muscle fatigue, disrupting the process of tissue respiration.
An increase in the level of transaminases (ALT and AST) in the blood always indicates greater than normal destruction of the cells of the corresponding organ, in our case the muscles.
Conclusion. Thus, the diagnosis of progressive muscular dystrophies is a complex task, the solution of which requires the creation and use of diagnostic algorithms.
The diagnosis of progressive Duchenne muscular dystrophy is based on clinical and genealogical history, clinical features of the disease (onset at 2-5 years, symmetrical atrophy of proximal muscle groups, their development in an ascending direction, pseudohypertrophy of the gastrocnemius muscles, gross somatic and neuroendocrine disorders, rapid malignant course of the disease ).
Rice. 1. Laboratory indicators in the examined children in a comparative aspect
Data from biochemical studies are also important (an increase in serum CPK levels is 71 times higher than normal against the background of increased ALT, AST and LDH), electromyography and pathomorphology, in which the primary muscular type of lesion is determined.
Diagnosis of Erb-Roth muscular dystrophy
- The diagnosis of the disease in question is based on a physical examination of patients, a study of the patient’s family history and subsequent analysis of the collected data.
- Genetic testing is carried out. Necessary for subsequent accurate determination of muscular dystrophy.
- Electroneuromyography.
- A biopsy of muscle tissue is performed with biochemical testing.
- A general blood test is given.
- A blood test for creatine phosphokinase is taken.
- Analysis of the patient's urine.
As for electromyography, it makes it possible to study not only the degree of neuromuscular transmission, but also to determine the level of direct muscle excitability, which is extremely important for the differential diagnosis of the disease with pathologies of neuropathic muscles.
What is limb-girdle muscular dystrophy?
Translator's note.
The name varies in different sources: limb-girdle MD and limb-girdle MD. Limb-girdle muscular dystrophy (LGMD) is not one common disease. This is a whole group of diseases that affect muscles, mainly located in the hips and shoulders.
The shoulder girdle is the bony structure that surrounds the shoulder region.
(Information from Wikipedia: the shoulder girdle (girdle of the upper limbs) is a set of bones (pairs of shoulder blades and collarbones) and muscles that provide support and movement of the upper (front) limbs.)
The pelvic girdle is a bony structure surrounding the hip area.
Collectively they are called “limb belts”. In PCMD, the muscles that are most damaged are those connected to the bones of these girdles.
The term " proximal " is also used to describe the muscles damaged in PCMD. Proximal muscles are muscles located close to the center of the body. Distal muscles are muscles that are further away from the center of the body (for example, muscles of the hands or feet). Distal muscles in PCMD are affected at later stages, but may retain their function.
As of the end of 2012, there are more than 20 different subtypes of PCMD. This is a complex and constantly evolving area of research.
Treatment of Erb-Roth dystrophy
It should be noted right away that I cannot restore the damaged gene, but...
Doctor Nikonov
The impact of my method is aimed at reducing the intensity of symptoms, slowing down the progression of the disease, increasing muscle strength, and restoring proper movement in all muscle groups.
I do not treat Erb-Roth muscular dystrophy. I am working on restoring normal muscle function . Therefore, I will not describe the treatment procedures used in hospitals and other rehabilitation centers in my article. Chances are you've tried them.
My article is for informational purposes only. I took the information from my observations and ten years of research and discoveries of scientists from all over the world.
Medical certificate
The disease is a polymorphic variant of hereditary myodystrophy. It differs from other types of pathology in its clinical picture, course and time of onset. The description of the disease was first presented by the German neurologist W. Erb in 1882. At the same time, V. Roth was working on this problem in Russia, which he later designated as “tabes muscularis.” It was after the names of the two scientists that the disease was named. In modern neurology, several of its names are used - progressive Erb-Roth muscular dystrophy, limb-girdle muscular dystrophy.
Pathology begins its development, as a rule, in childhood. However, the age at which symptoms first appear can range from 10 to 30 years. Men and women suffer equally from the manifestations of muscular dystrophy. Neurologists note that the disease, which began in childhood, progresses rapidly when compared with its course in adolescence and adulthood. In addition, in the second case it occurs in a mild form.
Treatment
The disease is incurable, since ways to eliminate the gene abnormality are only being studied. Therapeutic methods are aimed at slowing down the progress of Erb-Roth muscular dystrophy, relieving symptoms, and improving metabolism. The emphasis is on balanced nutrition, taking vitamin and mineral complexes, massage, exercise therapy, and physiotherapy.
What is prescribed for Erb-Roth muscular dystrophy:
- vitamins E, group B, D, C, A;
- adenosine triphosphoric acid;
- intramuscular injections of aloe extract;
- Metabolics - Actovegin, Riboxin, Thioctic acid;
- galantamine preparations;
- antioxidants, antihypoxants - trimetazidine agents;
- introduction of fetal stem cells;
- calcium chloride - for iontophoresis;
- Prozerin - for electrophoresis;
- 4th generation antidepressants with escitalopram, fluoxetine.
Reference! For Erb-Roth disease, they stopped using Retabolil and other nandrolone hormonal drugs. Clinical observations revealed an acceleration in the breakdown of muscle fibers.
With Erb-Roth muscular dystrophy, the patient should consume protein foods (meat, fish) and fresh fruits, vegetables, nutritional mixtures with amino acids, L-carnitine every day. Their substances are used by the body to build muscle fibers. It is important to drink mineral water (doctors select the type and daily intake individually). It is allowed to use folk remedies after consultation with a doctor.
To improve your health, the following are useful:
- lapacho tea - ant tree bark;
- infusion of rose hips;
- elecampane decoction;
- honey;
- pollen.
With Erba-Roth muscular dystrophy, exercise therapy, breathing exercises and massage are done daily. Swim at least once a week. Methods are used to strengthen and increase muscle volume, normalize cell nutrition, and microcirculation. This improves metabolism, maintains the ability to move, and removes fluid stagnation in organs and soft tissues.
Principles of exercise therapy for Erb-Roth myodystrophy:
- the load and exercises are selected by the doctor according to the state of health;
- the pace of execution and intensity are moderate or average;
- train all muscle groups;
- people with disabilities are helped to do exercises by loved ones;
- the duration of the workout should not exceed 20 minutes;
- a large set of exercises is divided into 2-3 times a day.
Maintenance massage is performed with light pressure for at least half an hour. Use kneading, stroking, rubbing, point, longitudinal movements. When caring for a disabled person, it is necessary to massage the chest area more often and more intensely to prevent the development of congestive pneumonia.
To stretch shortened muscles, an elongating massage is performed. For Erba-Roth muscular dystrophy, such a session is carried out for 20 minutes, pressing harder, using stretching, kneading, vibrating movements.
Complications are treated with etiological medications and methods depending on which organ is affected. For example, in the case of pneumonia, antibiotics are used; in case of respiratory failure, artificial ventilation is prescribed.
Prevention
Primary prevention includes refusing to marry a blood relative and following safety rules when working with harmful substances. People with confirmed heterozygous carriage should be aware of the risk of having children with muscular dystrophy. Doctors should monitor their child from infancy, and parents should do gymnastics and massage for the baby every day.
Important! Exercise therapy, nutritional therapy, massage and physiotherapy methods help slow down the progress of Erb-Roth muscular dystrophy at any age. Only prevention of complications is carried out.