Особенности течения мышечной дистрофии Ландузи-Дежерина
Нервно-мышечное заболевание характеризируется прогрессирующим поражением лицевой мускулатуры, плеч и лопаток. Также поражаются мышцы других частей тела, но значительно медленнее и менее выражено. Особенностью течения является несимметричность деструктивного процесса. Зачастую первыми атрофируются мышцы плечевого пояса, постепенно распространяясь на мимическую мускулатуру, приводя к амимичности лица, слабости и невозможности поднять руки.
В отличие от других миодистрофий, при миопатии Ландузи-Дежерина занятия спортом и физические нагрузки не замедляют патологический процесс, а наоборот ускоряют деструкцию мышечной ткани. В дальнейшем отмечается нарушение сердечной деятельности, может произойти потеря зрения и снизиться слух.
Впервые данную миопатию описали французские врачи Ландузи и Дежерин в 1884 году.
Известно, что диагноз является генетически обусловленным, с аутосомно-доминантным типом наследования. У большинства больных обнаруживаются генные мутации, но у некоторых пациентов генетические нарушения не выявляются.
Плече-лопаточно-лицевая миопатия Ландузи-Дежерина относится к редким патологиям, с частотой заболеваемости 1-1,5 на 100 000 населения. Из-за наследственной предрасположенности она часто выявляется у кровных родственников. Первые симптомы возникают еще в детском возрасте. Пик заболеваемости и выраженности клинических проявления – переходной возраст, период беременности и климактерический период.
Патологию продолжают изучать, но причина возникновения дефекта и развития атрофии не известна. Патогенетического лечения в настоящее время не существует, из-за этого болезнь продолжает относиться к тяжелым заболеваниям. Несмотря на то, что длительное время пациенты остаются трудоспособными и могут самостоятельно себя обслуживать, в большинстве случаев диагноз приводит к их инвалидизации.
Организации и сообщества, посвящённые ЛЛПМД. Регистры пациентов с ЛЛПМД
На сайте TREAT-NMD вы сможете найти подробную информацию о международных организациях, занимающихся лице-лопаточно-плечевой мышечной дистрофией Ландузи-Дежерина: https://www.treat-nmd.eu/FSHD/overview/. Также на этом сайте вы сможете найти международные регистры пациентов.
Почему нужно регистрироваться в реестре?
По мере того, как разрабатываются новые препараты, появляется необходимость их тестирования в клинических условиях, и иногда требуются годы, чтобы найти необходимое количество пациентов для исследований, поскольку мышечные дистрофии являются редким (орфанным) заболеванием.
Для этого в разных странах ведутся реестры (регистры) пациентов — базы данных по генетической и клинической информации о людях, страдающих нервно-мышечными заболеваниями и желающих ускорить процесс исследований. Реестр позволяет специалистам получить информацию о состоянии и количестве больных определённым заболеванием. Данная информация способствует развитию и улучшению стандартов лечения пациентов. Он используется, чтобы найти участников для проведения клинических испытаний, а также помочь специалистам получить больше информации о заболевании.
Причины развития
Первичные причины возникновения биохимических дефектов, приводящих к миопатии, неизвестны. Но, анализируя статистические данные, ученые выделяют несколько факторов развития миопатии:
- наследственный фактор – часто страдают члены одной семьи;
- генетическая предрасположенность – у многих выявляется дефект 4 хромосомы;
- вирусные заболевания у матери во время беременности;
- патология беременности у матери.
У некоторых пациентов миопатия возникает на фоне нормального здоровья без видимых на то причин. В таком случае диагноз классифицируют как идиопатическую миопатию.
Из-за отсутствия информации о начальных процессах развития патологии врачам практически не удается их остановить и восстановить утраченные функции организма.
Симптомы
Первые признаки заболевания возникают в 10-20 лет, когда отмечается чрезмерная мышечная слабость, быстрая утомляемость и медленное развитие мускулатуры плечевого пояса. Больным трудно поднимать руки, выполнять физические упражнения. Из-за атрофии мышечной ткани у них чрезмерно вступают лопатки, приобретая крыловидную форму, промежуток между ними расширен, грудная клетка уплощенная. Слабость мускулатуры способствует развитию сколиоза.
Дистрофия мышц плеча, грудной клетки и трапециевидной мышц приводит к развитию симптома свисающего плеча. Частым осложнением в таком случае являются вывихи плечевого сустава. У больных возникают трудности в повседневной жизни и в быту. Им сложно расчесывать волосы, умываться, чистить зубы. Из-за привычных ранее нагрузок они чрезмерно быстро устают, вплоть до невозможности их выполнения.
Прогрессируя, миопатия вовлекает в процесс лицевые мышцы. Лицо становится амимичным. Из-за поражения круговых мышц рта и глаз сложно либо невозможно зажмурить глаза, сжать губы. Это приводит к частым воспалениям глаз, травмированию роговицы.
Распространенной жалобой является нарушение речи – неразборчивая, медленная. Это значительно ухудшает качество жизни, особенно социальной жизни и трудовой деятельности. Также трудности возникают во время приема пищи. Больным тяжело есть твердую пищу, пережевывать ее и глотать.
Характерными признаками болезни являются: вывороченные губы «губы тапира», поперечная улыбка «лицо Джоконды», полированного лба.
В дальнейшем возможна прогрессирующая мышечная дистрофия в других частях тела. При поражении тканей ягодиц и бедра, мышечная слабость способствует чрезмерной утомляемости от ходьбы, которая может привести к хромоте. Поражение икроножных мышц сопровождается симптомом свисающей стопы, выраженной слабостью при ходьбе и невозможностью бегать.
В зависимости от того, какие симптомы более выражены и какова очередность их возникновения, выделяют следующие формы миопатии Ландузи-Дежерина:
- лицелопаточно-плечевая;
- лицелопаточная-плече-перонеальная;
- лицелопаточно-плече-ягоднично-бедренная;
- лицелопаточно-плече-ягоднично-бедренная-перонеальная;
- лицелопаточно-плече-перонеально- ягоднично-бедренная;
- инфантильная форма.
Наиболее тяжелой является инфантильная форма. Она возникает у детей до 5 лет. Характеризируется симметрическим парезом лицевых мышц, поражением плеч, грудной клетки, отсутствием сухожильных рефлексов. Из-за стремительно нарастающей слабости у детей возникает дыхательная недостаточность вплоть до необходимости в искусственной вентиляции легких.
Важно! Часто родители детей с чрезмерной мышечной слабостью стараются восстановить организм с помощью спортивных секций и физической нагрузки. В результате этого болезнь прогрессирует еще быстрее.
[править] Клиническая картина
В классическом варианте первые признаки заболевания начинают появляться в подростковом возрасте (около 10-17 лет). Обычно начальными проявлениями являются, такие симптомы как затруднение подъема рук над головой, расчесывания, становится трудно переносить привычные физические нагрузки. Визуально, становятся выраженными: выступающие «крыловидные» лопатки и сколиоз.
Атрофии и мышечная слабость локализуются в области мимической мускулатуры, лопаток и плеч. Сначала атрофии наблюдаются в плечевом поясе, с последующим распространением на лицо.[1]
При прогрессировании процесса грубо страдают круговые мышцы рта и глаз — не удается крепко зажмурить глаза и сжать губы. Вследствие атрофии лицо становится гипомимичным. Очень часто, больные сами отмечают изменение своей мимики, их речь постепенно становится неразборчивой. Патогномоничными симптомами являются, такие симптомы как поперечная улыбка («улыбка Джоконды»), вывороченные губы («губы тапира») и «полированный» лоб. Атрофии двуглавой и трехглавой мышц плеча, большой грудной, передней зубчатой, трапециевидной мышц, могут обусловливать возникновение симптомов свободных надплечий, «крыловидных» лопаток, появление широкого межлопаточного промежутка, уплощения грудной клетки и сколиоза. В ряде случаев атрофии могут распространяться на мышцы ног. При этом слабость наиболее заметна в группе малоберцовых мышц по свисающей стопе, но может присутствовать и в проксимальных отделах ног.
На ранних стадиях болезни мышечный тонус снижен в проксимальных группах мышц, глубокие рефлексы снижены преимущественно с двуглавой и трехглавой мышц плеча.
Необходимо отметить асимметричность атрофии, что является характерной клинической особенностью данной патологии. В некоторых случаях имеет место псевдогипертрофия мышц. Контрактуры и ретракции выражены умеренно. Кардиомиопатия бывает в редких случаях. При проведении ангиоретинографии могут выявляться аномалии сосудов сетчатки.
Во многих случаях при тяжелых глазных проявлениях находят телеангиэктазии, отек и отслойку сетчатки. Коагуляция телеангиэктазий предотвращает развитие слепоты. Может наблюдаться также и снижение слуха. Вышеперечисленные симптомы рассматриваются в качестве составляющей части фенотипических проявлений данной патологии.
Тип течения болезни в большинстве случаев относительно благоприятный
Методы диагностики
Несмотря на частое обнаружение генетической мутации во время генетического исследования, у некоторых больных с выраженной клинической картиной они отсутствуют. Из-за этого генетический анализ крови не является основным критерием заболевания. В большинстве случаев миопатию Ландузи-Дежерина диагностируют на основании характерных клинических проявлений, в первую очередь поражение типичных для болезни мышц.
Также дополнительно проводят следующие виды обследования:
- биохимический анализ крови с определением уровня креатинфосфокиназы;
- биопсию мышц;
- электронейромиографию.
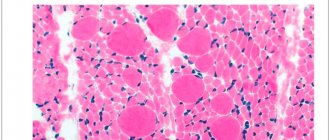
Значительное повышение уровня креатинфосфокиназы свидетельствует о прогрессирующей атрофии мышечной ткани. У больных отмечается слабая активность миоцитов. Для гистологического исследования производят забор образцов тканей с разных участков тела. При исследовании обнаруживаются характерные признаки атрофии. Дополнительно больные нуждаются в офтальмологическом обследовании и консультации невропатолога.
Важно! Течение некоторых болезней схоже с данной миопатией. Врачи обязательно проводят дифференциальную диагностику с неврологическими заболеваниями, аутоиммунными и патологиями соединительной ткани.
[править] Диагностика
Для уточнения диагноза используют биохимическое исследование с определением креатинфосфокиназы (КФК), электронейромиографию (ЭНМГ) и биопсию поврежденных мышц.
Уровень КФК может повышаться в 5-7раз, но в некоторых случаях содержание фермента нормальное. На ЭНМГ регистрируются как мипатические, так и денервационные потенциалы.
При проведении гистологических исследований во многих мышцах конечностей выявляют минимальные изменения, наибольшее число патологических признаков отмечается в надлопаточных мышцах, где обнаруживаются явления прогрессирующей дегенерации.
Лечение
Заболевание относится к неизлечимым диагнозам. Патогенетическое лечение отсутствует. Также достаточно сложно остановить быстропрогрессирующую атрофию. Лечение миопатии Ландузи-Дежерина направленно в первую очередь на замедление дистрофических изменений, приводящих к невозможности самостоятельно передвигаться и обслуживать себя, восстановление утраченных функций и общее улучшение состояния пациента. Медикаментозная терапия основана на применении медикаментов, поддерживающих миоциты, улучшающих обменные процессы в них, и благотворно воздействующих на сердце. Наиболее часто назначают такие препараты: АТФ-лонг, Милдронат, Метион, витамин Е, Цитофлавин, кокарбоксилаза.
Также для улучшения состояния значение имеют следующие факторы:
- питание – рекомендуется увеличить количество белков и сократить жиры;
- физиопроцедуры – больным показан массаж, электрофорез и другие процедуры;
- лечебная физкультура – умеренные физические нагрузки и специальный комплекс необходим для предупреждения развития сколиоза и контрактур.
Учитывая, что миопатия зачастую возникает в детском возрасте, особое внимание необходимо уделять психическому состоянию пациентов. Часто патология приводит к депрессивным расстройствам, нервным срывам и нарушениям эмоциональной сферой. Чтобы предотвратить психологические нарушения и улучшить эмоциональное состояние пациентов, им необходима помощь психолога или психотерапевта.
Для социализации необходимо помочь больным с выбором профессии, чтобы сохранить их работоспособность.
Посмотрите видео по теме статьи!
Возможные осложнения
Несмотря на тщательное изучение, миопатия Ландузи-Дежерина продолжает относиться к тяжелым патологиям. Во многом прогноз зависит от особенностей течения и скорости развития патологического процесса. Гипомимия и амимия не только ограничивает физические возможности, но и является причиной психологического дискомфорта и трудностей с социализацией.
Прогрессирование и вовлечение в патологический процесс других частей тела приводит к значительным ограничениям, вплоть до невозможности передвигаться.
В некоторых случаях при поражении круговых мышц глаза и частых воспалительных процессах у пациентов ухудшается зрение вплоть до слепоты.
Зачастую, выполнение рекомендаций врача помогает сохранить продолжительность жизни, но при крайне тяжелой форме заболевания возможен летальный исход из-за тяжелой дыхательной недостаточности.
Что нужно запомнить?
- Миопатия Ландузи-Дежерина характеризируется прогрессирующей атрофией мышц лица, плеч и лопаток.
- Основной причиной заболевания является генетическая предрасположенность.
- Признаками патологии является гипомимичность и амимичность, мышечная слабость и потеря мышечной ткани.
- Лечение направлено на торможение развития болезни и предотвращение ее прогресса.
- Мышечная дистрофия приводит к инвалидизации пациента из-за выраженных ограничений его возможностей.
- Прогноз на выздоровление неблагоприятный, так как болезнь неизлечима.
Литература
- Асанов А. Ю. Основы генетики и наследственные нарушения развития у детей: Учеб. пособие для студентов вузов. — М.: Академия, 2003. — С. 216.
- Бадалян Л. А. Детская неврология: учеб. пособие. 4-е изд. — М.: МЕДпресс информ, 2021. — 608 с.
- Влодавец Д. В., Сухоруков В. С., Харламов Д. А., Белоусова Е. Д. Способ лечения врожденных структурных миопатий и врожденных мышечных дистрофий путем коррекции вторичных митохондриальных изменений: Автореф. дис. … к. м. н. — М., 2009. — 28 с.
- Гринио Л. П. Атлас нервно-мышечных болезней. — М.: АНС, 2004. —167 с.
- Ерохина В. А. Реабилитация детей с наследственными миопатиями // Вестник Московского Университета МВД России. — 2015. — № 12. — С. 304—308.
- Arvanitidis A., Henriksen K., Karsdal M. A., Nedergaard A. Neo-epitope Peptides as Biomarkers of Disease Progression for Muscular Dystrophies and Other Myopathies // J Neuromuscul Dis. — 2016. — № 30. — Р. 333—346.
- Carroll M. B., Newkirk M. R., Sumner N. S. Necrotizing Autoimmune Myopathy: A Unique Subset of Idiopathic Inflammatory Myopathy // J Clin Rheumatol. — 2021. — № 22. — Р. 376—80.
- Inoue M., Nishino I. Diagnosis of Idiopathic Inflammatory Myopathy: A Muscle Pathology Perspective // Brain Nerve. — 2021. — № 68. — Р. 1431—1441.
Лечение миодистрофий
Лечение направлено на предотвращение или уменьшение выраженности поражений суставов, позвоночника и других последствий основного заболевания. Главная задача — обеспечить больному мобильность максимально возможный период времени. Для этого назначают лекарственные препараты, физиотерапию, иногда — хирургическое лечение3,4.
Справочная литература
- Muscular Dystrophy Information Page. NIH (дата обращения 20.07.2019). URL: https://www.ninds.nih.gov/Disorders/All-Disorders/Muscular-Dystrophy-Information-Page.
- Никитин С. С. и др. Болезнь Помпе с поздним началом: первое клиническое описание в России //Нервно-мышечные болезни – 2014. – № 1.
- Жданова Е. Б., Харламов Д. А., Белоусова Е. Д. Соматические нарушения при прогрессирующей мышечной дистрофии Дюшенна //Российский вестник перинатологии и педиатрии – 2011. – Т. 56. – № 5.
- Мышечная дистрофия Дюшенна и мышечная дистрофия Беккера. Справочник MSD (дата обращения 20.07.2019).
- Белозеров Ю. М. и др. Прогрессирующая мышечная дистрофия Эмери-Дрейфусса //Альманах клинической медицины – 2001. – № 4.
- Умаханова З. Р., Жилина С. С., Мутовин Г. Р. Клинический полиморфизм прогрессирующей мышечной дистрофии Эрба-Рота //Журнал неврологии и психиатрии им. CC Корсакова – 2005. – Т. 105. – № 9. – С. 48-51.
- Кириллова Л. Г. и др. Лицелопаточно-плечевая миодистрофия Ландузи-Дежерина в клинике нейропедиатрии //Здоровье ребенка – 2011. – № 1.
GZEA.PD.18.09.0435m