Лиссэнцефалией (буквально «гладкий мозг») называется весьма большая по объему группа нарушений в развитии головного мозга, которая характеризуются частичным отсутствием или же плохим развитием извилин, расположенных в полушариях головного мозга.
В итоге поверхность мозга становится удивительно гладкой. Этот дефект начинает проявляться еще с 12 недели развития плода, и заканчивает формироваться спустя 2 недели, и вызван он нарушенным процессом передвижения нейронов в этот период.
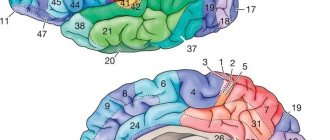
Головной мозг, в нормальном состоянии у каждого здорового человека, имеет множество складок и борозд. У плода с этим отклонением, структура развивается не своеобразным способом, у таких пациентов складки и борозды либо частично отсутствуют, либо их вовсе нет, и произошедшие изменения уже не обратимы.
Точно так же, как и многие другие дефекты в развитии головного мозга, лиссэнцефалия берет под контроль большой спектр фенотипов, они могут колебаться в соответствии с тяжестью от переменной агирии, либо до полной.
Лиссэнцефалия, шизэнцефалия, порэнцефалия: причины, симптомы, прогноз
Сглаживание извилин коры головного мозга (агирия) формируется внутриутробно. Патогенетическим механизмом развития нозологии является нарушение эмбриогенеза, обусловленного патологией распространения из первичной нервной трубки нейробластов. Сглаживание поверхности сопровождается снижением умственной, интеллектуальной активности.
Сопутствующие неврологические расстройства постепенно нарастают. Заболевание приводит к летальному исходу еще в грудном возрасте.
Отсутствие мозговых извилин – исключает оптимальное функционирование второй сигнальной системы, которая существует только у человека. Складчатость повышает функциональную поверхность мозга.
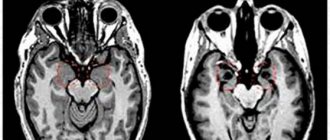
Даже раннее выявление с помощью МРТ головного мозга не позволяет спасти жизнь ребенку с лиссэнцефалией первого или второго типа.
На протяжении беременности врожденные пороки развития церебральных структур определяются ультразвуковым обследованием (УЗИ). Достоверность процедуры – 60%, поэтому иногда после МРТ выявляются врожденные пороки развития мозга, хотя внутриутробный мониторинг патологии не определяет патологических состояний.
Пример заключения магнитно-резонансной томографии:
- Билатеральная вентрикуломегалия;
- Увеличение ликворных пространств;
- Сглаженность церебральных извилин;
- Неполное отсутствие складок теменных и лобно-височных областей;
- Расширение боковых желудочков.
Нозология нередко сочетается со стигмами церебрального дисэмбриогенеза – макроцефалией, агирией, микроцефалией.
Медицинские источники утверждают, что патологию может обнаружить УЗИ после двадцать шестой недели беременности.
МРТ: норма и лиссэнцефалия
Лиссенцефалия 2 типа имеет альтернативное название «синдром Норман-Робертс». Канадские ученые, открывшие заболевание, считают основной причиной формирования заболевания мутацию гена «RELN».
Участок ДНК, отвечающий за нозологию, кодирует образование протеина «рилина». Синдром характеризуется множеством клинических изменений:
- Лимфедема (лимфатический отек);
- Эпилептические приступы;
- Покатость лба;
- Утолщение церебральной коры;
- Агирия (сглаженность мозговых борозд);
- Макроцефалия;
- Микроцефалия.
Перинатальное УЗИ обнаруживает большую часть случаев болезни.
Причины лиссэнцефалии
Выделяют различные виды мутационных изменений с разной локализацией, приводящие к данной патологии. Факторами риска, помимо наследственной предрасположенности, являются:
- инфекционные болезни женщины в период вынашивания ребенка;
- влияние химических и ядовитых веществ;
- работа во вредных производственных условиях;
- воздействие тератогенных агентов;
- прием медикаментов с побочными негативными эффектами по отношению к плоду.
Среди мутаций наибольшее значение имеют делеции, то есть, выпадение фрагмента хромосомы. В некоторых клинических случаях болезнь проявляется не только нарушением закладки и образования структур коры мозговых полушарий, но и патологическими изменениями со стороны половой, сердечно-сосудистой, пищеварительной, мочевыделительной систем.
Причины возникновения лиссенцефалии
Формирование внутримозговой коры у плода проходит несколько стадий развития. Вначале происходит деление нейронов. Постепенно нервные клетки мигрируют из первоначального матрикса в места постоянного расположения. Остальные этапы эмбриогенеза сопровождаются специализацией клеточных пластов, отвечающих за анатомические структуры.
Сложный процесс образования мозга должен проходить под строгим контролем биохимических механизмов. Любое внешнее вмешательство нарушает внутриутробное развитие.
Основные причины возникновения лиссэнцефалии:
- Внутриутробная гипоксия (кислородное голодание);
- Вирусные инфекции;
- Хромосомные аномалии (дефекты формирования протеина рилина в 7-ой хромосоме).
Внутрисемейные случаи передачи заболевания достаточно редки. Если встречается нозология у нескольких членов семьи, требуется генетическое консультирование.
Средняя распространенность нозологии первого типа – 11% на миллион новорожденных детей. Большинство ситуаций провоцируется генетическими дефектами гена LIS1. На втором месте встречается гипоплазия мозжечка из-за дефекта гена TUBA1A.
После наследственных аномалий на втором месте – вирусное инфицирование в первом триместре беременности.
Что такое микроделеционный синдром?
Самые незначительные изменения (они же мутации) называются точечными. Их появление влияет на считанные единицы генов. В некоторых случаях нарушение относится вообще к одному единственному гену. Однако если он обеспечивал выработку важного белка, последствия для всего организма могут быть очень серьезными. Подобные патологические изменения относятся к группе микроделеционных синдромов.
Каждое такое заболевание обусловлено небольшим изменением генетического материала, которое происходит в строго определенном месте. Точный механизм возникновения подобных нарушений на сегодняшний день не установлен, что не мешает ученым заниматься исследованием их воздействия на организм.
Так, было выяснено, что развитие синдрома в таком случае может происходить несколькими различными вариантами. В частности, ряд заболеваний характеризуется участием онкогенов. В других случаях на воздействие непосредственно самой делеции накладывается эффект хромосомного импринтинга и возможные однородительские дисомии.
Частота возникновения большей части микроделеционных синдромов крайне невелика: порядка 1 случая на 50-100 тысяч новорожденных. Набор клинических признаков обычно выражается отчетливо. Для того чтобы поставить диагноз, бывает достаточно лишь совокупности симптомов. Однако при таком подходе невозможно точно прогнозировать здоровье потомков, поэтому зачастую наряду с проверкой обычных признаков производится молекулярно-генетическая диагностика пробанда и его родственников (обычно родители, в некоторых случаях также требуется анализ генотипа братьев, сестер, теть, дядь и так далее).
Патологические проявления сильно отличаются. В частности, их проявление определяется тем, насколько большой участок генетического материала был утрачен в результате делеции. Кроме того, в ряде случаев играет роль то, от кого из родителей была получена мутация (влияние хромосомного импринтинга). Хорошей иллюстрацией последней ситуации является пара синдромов Прадера-Вилли и Ангельмана. Они оба обусловлены наличием делеции в 15 хромосоме. Однако из-за различного механизма действия при передаче от разных родителей клиническая картина этих заболеваний значительно отличается.
Основные виды лиссенцефалии
Встречается свыше двадцати морфологических форм заболевания:
- Классическая форма (I типа) с мутацией LIS1, синдромом Миллера-Дикера;
- Форма с наследственным дефектом DCX;
- Изолированная разновидность без мутаций;
- Агенезия мозолистого тела при аномалии Х-хромосомы;
- Гипоплазия мозжечка с нарушением кодирования рилина (синдром Нормана-Робертса);
- Микролиссэнцефалия;
- Булыжниковый вид с синдромом Фукуямы, Уокера-Варбурга, мышечно-глазо-мозговой формой.
Мышечная дистрофия с олигофренией Фукуяма передается аутосомно-рецессивным способом. Наследственная миотония имеет прогрессирующее течение.
Какие бывают типы и виды лиссэнцефалии
Классификация заболевания имеет много пробелов из-за большого количества разнообразных мутаций различной локализации, но на данный момент она включает в себя пять видов — ставится диагноз лиссэнцефалия 1-го, 2-го, 3-го, 4-го, 5-го типа, а именно:
- проявляется аномальной складчатостью поверхности головного мозга. В эту группу отнесены пороки, которые передаются сцеплено с полом, мутации в гене LSI1, а также болезни с аналогичными проявлениями не выясненной этиологии;
- передается с Х хромосомой. Помимо нарушений анатомо-физиологического строения коры полушарий, наблюдаются расстройства со стороны половой системы и панкреаса, центра терморегуляции;
- определяется частичное недоразвитие или полное отсутствие мозжечка;
- все клинические признаки дополняются маленьким размером головного мозга;
- болезни вызваны мутацией в девятой паре хромосом.
Особенности шизэнцефалии, порэнцефалии, голопрозэнцефалии
Билатеральная расщелина церебральных полушарий формируется на этапе эмбриогенеза. Морфологическая структура образования бывает разомкнутой или сомкнутой.
Односторонняя локализация патологии на МРТ снимках может напоминать кисту. Вдоль расщелины расположены патологические участки церебральной паренхимы – микрогирия, зоны повышенной повторной активности, спастические параличи.
Порэнцефалия характеризуется образованием кистозных полостей внутри церебральной паренхимы. Причины возникновения патологии – кровоизлияние внутрь головного мозга, инфаркты, инсульты. Типичная локализация кисты – сильвиева борозда.
В редких случаях порэнцефалия характеризуется образованием аномальных ходов между кистозной полостью и желудочковыми пространствами. Нозология нередко характеризуется другими стигмами дисэмбриогенеза:
- Микроцефалия;
- Энцефалоцеле;
- Агирия.
У ребенка с пороком развития возникают неврологические расстройства:
- Олигофрения (умственная отсталость);
- Приступы эпилепсии;
- Атрофические изменения дисков зрительного нерва;
- Тетрапарез.
Осложняют клиническую картину сопутствующие изменения:
- Артериальные и венозные кровоизлияния;
- Внутричерепная гипертензия (при нарушении циркуляции ликвора);
- Церебральные инфаркты;
- Гемипарезы;
- Очаговые эпилептические очаги.
Псевдопорэнцефалические кисты нередко имеют унилатеральное (одностороннее) расположение. Нередко сочетаются с пороками центральной нервной системы, дефектами клеточной миграции.
Шизэнцефалия на МРТ
Голопрозэнцефалия – что это такое
Заболевание возникает по причине патологического разделения химического вещества прозэнцефалона.
Классификация нозологии по степени тяжести:
- Лобарная;
- Полулобарная;
- Алобарная.
Самая тяжелая – последняя форма. При ней наблюдается ряд врожденных аномалий:
- Недоразвитие межчелюстной кости;
- Синофтальм;
- Цебоцефалия.
На фоне множественных аномалий части лица трудно различимы. Сопутствующие пороки нейрональной миграции:
- Синостоз (сращение нервных ганглиев);
- Наличие одного церебрального желудочка.
Этиологические механизмы наследственных дефектов не установлены. Голопрозэнцефалия в большинстве случаев завершается смертельным исходом, но неполная форма может приводить к инвалидности.
Синдром Прадера-Вилли
Это заболевание определяется той же самой генетической мутацией, что и для синдрома Ангельмана. Отличие состоит в том, что при этом нарушение наследственного материала получается со стороны отца. Кариотип соответствует нормальному (46XX или 46XY). По распространенности (1 случай на 12-15 тысяч новорожденных) примерно совпадает с распространенностью синдрома Ангельмана.
Характерными признаками синдрома Прадера-Вилли являются следующие симптомы:
· в пренатальный период малая подвижность плода;
· часто встречается неверное положение плода;
· возможна дисплазия тазобедреных суставов;
· к двум годам может проявиться склонность много есть (больше нормы), что приводит к ожирению;
· низкий мышечный тонус (гипотонус), сочетающийся с нарушенной координацией движений;
· стопы и кисти обычно маленькие, кроме того характерен невысокий рост;
· формирование косоглазия и сколиоза;
· отмечают повышенную сонливость;
· плотность костей находится на более низком уровне, чем у здоровых людей;
· слюна густая, обычно состояние зубов плохое;
· недостаточная функция половых желез, вызывающая в итоге бесплодие;
· позднее по сравнению со сверстниками половое созревание;
· больные позже учатся говорить, отстают в психическом развитии;
· внешние признаки включают выраженную переносицу, узкий и высокий лоб, миндалевидную форму глаз, узкие губы.
В большинстве случаев у человека с мутацией насчитывается от одного до пяти признаков заболевания.
Диагностика заболевания проводится путем молекулярно-генетического тестирования, на которое направляются дети с пониженным мышечным тонусом. Зачастую вместо верного диагноза определяется более распространенный «синдром Дауна». Опытный генетик, достаточно часто встречающийся с проявлениями синдрома Прадера-Вилли способен диагностировать его по комплексу внешних признаков.
Симптомы лиссэнцефалии
Клинические проявления нозологии определяются сразу после рождения ребенка. Отмечаются не только генетические дефекты, но и нарушения глотания у малыша, повышенный мышечный тонус, отсталость в развитии от сверстников.
Симптомы лиссэнцефалии у ребенка 2-5 месяцев:
- Олигофрения;
- Нарушение умственного развития;
- Миоклония;
- Увеличение артериального давления.
Вначале умственная отсталость не прослеживается. После первого года жизни наблюдаются расстройства психоречевых навыков. Дети раздражительны, тревожны, мышечная активность снижена.
При сравнении со сверстниками наблюдается значительное отставание в развитии, формировании мышечной системы, иннервации внутренних органов, малого таза.
При легком течении заболевания ребенок может дожить до подросткового возраста. Внешние аномалии ограничивают социального общения. Больные редко доживают до восемнадцати лет, но существует около двадцати разновидностей патологии. Проявления первого типа:
- Макрогирия;
- Шизэнцефалия;
- Микроцефалия;
- Четырехслойное строение коры;
- Гипоплазия моста;
- Недоразвитие мозжечка.
Определить лиссэнцефалию при отсутствии положительных результатов внутриутробного УЗИ сложно. Клиническая картина возникает после рождения. Мышечные параличи, судороги появляются сразу или на протяжении первого года.
В большинстве случаев установить диагноз лиссэнцефалии поможет ультразвуковое обследование. Врач может назначить магнитно-резонансную томографию беременной женщине и ребенку. МРТ является безвредной, но в первом триместре не проводится из-за отсутствия практической информации о влиянии магнитного поля на плод.
Синдром Ангельмана
При синдроме Ангельмана развивается характерный набор патологических изменений. В частности, отмечается задержка психологического развития, сопровождающаяся проблемами со сном, частыми хаотическими движениям (больше руками), постоянными улыбками и смехом.
Патология развивается при отсутствии некоторых генов, расположенных на 15 хромосоме. При этом обязательным условием является передача мутантной копии гена от матери. Если поврежденная хромосома будет унаследована от отца, то разовьется синдром Прадера-Вилли. Кариотип обычно нормальный (46XX и 46XY для девочек и мальчиков соответственно). Различные независимые исследования указывают на связь болезни с геном UBE3A, который в норме обеспечивает выработку ферментного компонента в сложной системе деградации белков.
Частота появление синдрома составляет примерно 1 случай на 10-20 тысяч новорожденных (показатели отличаются у различных ученых).
Характерными особенностями больных с синдромом Ангельмана являются следующие признаки:
· проблемы с питанием, начинающиеся еще во время грудного вскармливания, поскольку дети плохо набирают вес (распространенность признака порядка 75 процентов);
· заторможенное развитие навыков общей моторики, то есть дети позже других начинают сидеть и ходить;
· для всех детей характерны нарушения речевого развития;
· больные обычно понимают больше, чем в состоянии выразить при помощи ограниченного словарного запаса;
· часто заболевание сопровождается дефицитом внимания и гиперактивностью;
· проблемы с обучением в обычной школе;
· у 80% заболевших развивается эпилепсия, сопровождающаяся заметными на электроэнцефалографии нарушениями; ученые полагают, что заболевание эпилепсией носит вторичный (симптоматический) характер.
· выполнение необычных движений, к которым относятся произвольные хаотические движения конечностями, мелкий тремор;
· возникновение приступов смеха при отсутствии видимых причин;
· характерная ходьба на негнущихся ногах, из-за которой возникло сравнение с марионетками;
· уменьшенная по сравнению со средними размерами голова, часто с уплощенным затылком;
· в некоторых случаях встречаются своеобразные запоминающиеся черты лица – широкий рот с редко расположенными зубами, выдвинутый вперед подбородок с выпущенным наружу языком;
· различные нарушения сна;
· примерно в 40 процентах случаев развивается косоглазие;
· порядка 10% больных также страдает от искривления позвоночника;
· высокие температуры воспринимаются с повышенной чувствительностью;
· наибольшего комфорта обычно достигают в воде (к примеру, в ванной)
Как правило, синдром определяется при помощи методов молекулярно-генетической диагностики по 15 хромосоме. Показанием к проведению тестирования для новорожденного является пониженный мышечный тонус (гипотонус), заметное отставание в развитии речи и мелкой моторики. Кроме того, на заболевание могут указывать мелкий тремор, порывистые беспорядочные движения, передвижение на негнущихся ногах.
Анализ может проводиться через флуоресцентную гибридизацию in situ, метилированием ДНК в области 15q11-q13. Также можно проверить мутации в импринтинговом центре и в гене UBE3A.
Поскольку заболевание обусловлено генетическим нарушением, адекватного и действенного способа лечения для него не имеется. Выполнение лечебных мероприятий, таких, как массаж для больных с гипотонусом, позволяет повысить качество жизни.
Первые признаки лиссэнцефалии у новорожденных
Раннее выявление нозологии помогает продлить жизнь малышу, но спасти от смерти не удается. Первые признаки у новорожденных:
- Небольшая голова;
- Апатия, вялость;
- Широкое межглазное расстояние;
- Эпилептический судорожный синдром;
- Гипертонус мускулатуры;
- Увеличение ширины между легкими и почками;
- Активация патологических рефлексов;
- Покатая лобная часть.
На протяжении первого года жизни присоединяются другие проявления – речевые расстройства, недержание мочи, учащение дыхания и сердцебиения.
Синдром летального птеригума (Нормана-Робертса) возникает при лиссэнцефалии первого типа. К вышеописанным проявлениям присоединяется ряд проявлений:
- Сложности сидения;
- Невозможность удержания малышом вертикальной позиции;
- Появление генерализованных подергиваний мускулатуры;
- Аномалии краниофациальной области (широкие глаза, покатый лоб, бугры на затылке);
- Глазной нистагм;
- Снижение мышечного тонуса.
Мозжечковая атаксия характеризуется нарушением координации. Ребенок не может удержать равновесия, поддерживать вертикальную позицию. Родители вначале обращают внимание на сложности посадки, сохранения горизонтального положения.
Диагностика лиссэнцефалии
Устанавливается диагноз сразу после рождения с помощью УЗИ, КТ и МРТ. Внутриутробно обнаружить проявления можно с 20 недели. Выявление малейших нарушений развития головного мозга плода требует дополнительной диагностики. В зависимости от степени повреждений принимается решение о возможности прерывания беременности. При подозрении на наследственные формы проводится генетическое консультирование всех членов семьи.
Поздняя верификация патологии требуется постоянного консервативного лечения. Терапия тяжелых форм проводится в стационаре под контролем квалифицированных врачей.
Основные виды диагностики лиссэнцефалии:
- Магнитно-резонансная томография (МРТ головы) показывает мягкие ткани. Исследование обнаруживает церебральные аномалии, кисты, скопления жидкости, воспалительные очаги;
- Внутриутробное УЗИ головы выполняется на 22-27 неделе, когда прослеживается процесс образования борозд;
- Компьютерная томография (КТ) помогает верифицировать изменения серого и белого вещества, дополнительные твердые образования. Обследование приводит к радиационному облучению тканей, поэтому детям выполняется по строгим показаниям;
- Электроэнцефалография (ЭЭГ) верифицирует очаги повышенной мозговой активности, участки гиперактивности паренхимы.
Самое достоверное исследование – МРТ при лиссэнцефалии у новорожденных, позволяющее определить самые мельчайшие аномалии.
Постановка диагноза и возможности медицины
Обычно в таком случае диагноз ставится при рождении или немного позже, после проведения УЗИ и исследования результатов КТ или МРТ.
Но врачи могут заподозрить наличие патологии еще во время беременности, ведь они регулярно проводят УЗИ плода, и постоянно изучают его состояние, в частности и развитие головного мозга.
Если врач заподозрит лиссэнцефалию, кроме УЗИ нужно будет проводить еще несколько методов диагностики, например, анализ на явность мутирующих генов и ЯРМ-томографию.
Обнаружить данную патологию можно не раньше, чем на 20 неделе беременности, поскольку поверхность мозга до этого времени, и без каких-либо нарушений, является гладкой. Именно после данного периода, можно заметить отклонения в развитии плода.
Вылечить данный порок невозможно, есть возможность симптоматического лечения, и оно зависит от стадии развития и места расположения дефектов. Необходимо поддерживать постоянный уход.
Для людей с гидроцефалией обычно делают шунтирование, а для контроля судорог принимаются специальные лекарственные препараты.