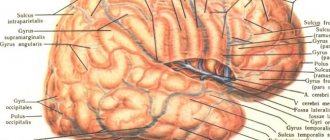
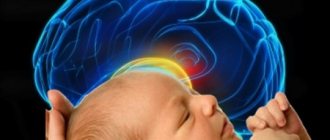
Lissencephaly (literally “smooth brain”) is a very large group of disorders in the development of the brain, which is characterized by the partial absence or poor development of convolutions located in the cerebral hemispheres.
As a result, the surface of the brain becomes surprisingly smooth. This defect begins to appear as early as the 12th week of fetal development, and finishes forming after 2 weeks, and it is caused by a disrupted process of neuron movement during this period.
The brain, in normal condition in every healthy person, has many folds and grooves. In a fetus with this deviation, the structure does not develop in a unique way; in such patients, folds and grooves are either partially absent or not at all, and the changes that have occurred are no longer reversible.
Just like many other defects in brain development, lissencephaly controls a wide range of phenotypes that can range in severity from variable agyria to complete agyria.
Lissencephaly, schizencephaly, porencephaly: causes, symptoms, prognosis
Smoothing of the convolutions of the cerebral cortex (agyria) is formed in utero.
The pathogenetic mechanism for the development of the nosology is a violation of embryogenesis caused by the pathology of the spread of neuroblasts from the primary neural tube. Smoothing of the surface is accompanied by a decrease in mental and intellectual activity. Concomitant neurological disorders gradually increase. The disease is fatal in infancy.
The absence of cerebral convolutions excludes the optimal functioning of the second signaling system, which exists only in humans. Folding increases the functional surface of the brain.
Even early detection using brain MRI cannot save the life of a child with type 1 or type 2 lissencephaly.
During pregnancy, congenital malformations of cerebral structures are determined by ultrasound examination (ultrasound). The reliability of the procedure is 60%, so sometimes congenital malformations of the brain are detected after MRI, although intrauterine monitoring of pathology does not determine pathological conditions.
An example of a magnetic resonance imaging report:
- Bilateral ventriculomegaly;
- Increased liquor spaces;
- Smoothness of the cerebral convolutions;
- Incomplete absence of folds in the parietal and frontotemporal regions;
- Dilatation of the lateral ventricles.
The nosology is often combined with the stigmas of cerebral dysembryogenesis - macrocephaly, agyria, microcephaly.
Medical sources claim that pathology can be detected by ultrasound after the twenty-sixth week of pregnancy.
MRI: normal and lissencephaly
Lissencephaly type 2 is alternatively called Norman-Roberts syndrome. Canadian scientists who discovered the disease consider the mutation of the “RELN” gene to be the main cause of the disease.
The DNA region responsible for nosology encodes the formation of the “Reelin” protein. The syndrome is characterized by many clinical changes:
- Lymphedema (lymphedema);
- Epileptic seizures;
- Sloping forehead;
- Thickening of the cerebral cortex;
- Agyria (smoothness of the cerebral sulci);
- Macrocephaly;
- Microcephaly.
Perinatal ultrasound detects most cases of the disease.
Causes of lissencephaly
There are various types of mutational changes with different localizations that lead to this pathology. Risk factors, in addition to hereditary predisposition, are:
- infectious diseases of women during pregnancy;
- influence of chemicals and toxic substances;
- work in hazardous production conditions;
- exposure to teratogenic agents;
- taking medications with negative side effects on the fetus.
Among mutations, deletions are the most important, that is, the loss of a chromosome fragment. In some clinical cases, the disease manifests itself not only as a violation of the formation and formation of structures of the cerebral cortex, but also by pathological changes in the reproductive, cardiovascular, digestive, and urinary systems.
Causes of lissencephaly
The formation of the intracerebral cortex in the fetus goes through several stages of development. First, neurons divide. Gradually, nerve cells migrate from the original matrix to their permanent locations. The remaining stages of embryogenesis are accompanied by the specialization of cell layers responsible for anatomical structures.
The complex process of brain formation must take place under the strict control of biochemical mechanisms. Any external intervention disrupts intrauterine development.
The main causes of lissencephaly:
- Intrauterine hypoxia (oxygen starvation);
- Viral infections;
- Chromosomal abnormalities (defects in the formation of the Reelin protein on chromosome 7).
Intrafamily cases of transmission of the disease are quite rare. If the nosology occurs in several family members, genetic counseling is required.
The average prevalence of type 1 nosology is 11% per million newborns. Most situations are caused by genetic defects of the LIS1 gene. In second place is cerebellar hypoplasia due to a defect in the TUBA1A gene.
After hereditary anomalies, viral infection in the first trimester of pregnancy is in second place.
What is microdeletion syndrome?
The most minor changes (aka mutations) are called point changes. Their appearance affects a few units of genes. In some cases, the disorder relates to just one gene. However, if it provided the production of an important protein, the consequences for the entire body could be very serious. Such pathological changes belong to the group of microdeletion syndromes.
Each such disease is caused by a small change in genetic material that occurs in a strictly defined location. The exact mechanism of occurrence of such disorders has not been established to date, which does not prevent scientists from studying their effects on the body.
Thus, it was found that the development of the syndrome in this case can occur in several different ways. In particular, a number of diseases are characterized by the participation of oncogenes. In other cases, the effect of the deletion itself is superimposed by the effect of chromosomal imprinting and possible uniparental disomies.
The incidence of most microdeletion syndromes is extremely low: about 1 case per 50-100 thousand newborns. A set of clinical signs is usually expressed clearly. In order to make a diagnosis, only a combination of symptoms is enough. However, with this approach it is impossible to accurately predict the health of the offspring, so often, along with checking the usual signs, a molecular genetic diagnosis of the proband and his relatives (usually parents, in some cases also requires analysis of the genotype of brothers, sisters, aunts, uncles, and so on) is carried out.
Pathological manifestations vary greatly. In particular, their manifestation is determined by how much of the genetic material was lost as a result of the deletion. In addition, in some cases, the role from which parent the mutation was received (the influence of chromosomal imprinting) plays a role. A good illustration of the latter situation is the pair of Prader-Willi and Angelman syndromes. They are both caused by the presence of a deletion on chromosome 15. However, due to the different mechanism of action when transmitted from different parents, the clinical picture of these diseases differs significantly.
Main types of lissencephaly
There are over twenty morphological forms of the disease:
- Classic form (type I) with LIS1 mutation, Miller-Dieker syndrome;
- Form with hereditary defect DCX;
- Isolated variety without mutations;
- Agenesis of the corpus callosum due to an abnormality of the X chromosome;
- Cerebellar hypoplasia with reelin coding disorder (Norman-Roberts syndrome);
- Microlissencephaly;
- Cobblestone appearance with Fukuyama syndrome, Walker-Warburg syndrome, musculocular-brain form.
Muscular dystrophy with Fukuyama mental retardation is transmitted in an autosomal recessive manner. Hereditary myotonia has a progressive course.
What are the types and types of lissencephaly?
The classification of the disease has many gaps due to the large number of different mutations of different localization, but at the moment it includes five types - the diagnosis is lissencephaly of the 1st, 2nd, 3rd, 4th, 5th type, namely:
- manifested by abnormal folding of the surface of the brain. This group includes defects that are transmitted in a sex-linked manner, mutations in the LSI1 gene, as well as diseases with similar manifestations of unknown etiology;
- transmitted on the X chromosome. In addition to disorders of the anatomical and physiological structure of the cerebral cortex, disorders of the reproductive system and pancreas, the center of thermoregulation, are observed;
- partial underdevelopment or complete absence of the cerebellum is determined;
- all clinical signs are complemented by a small brain size;
- diseases are caused by a mutation in the ninth pair of chromosomes.
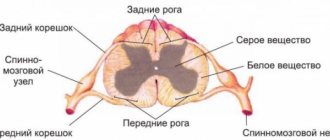
Features of schizencephaly, porencephaly, holoprosencephaly
Bilateral cleft of the cerebral hemispheres is formed at the stage of embryogenesis. The morphological structure of the formation can be open or closed.
Unilateral localization of pathology on MRI images may resemble a cyst. Along the cleft there are pathological areas of cerebral parenchyma - microgyria, zones of increased repeated activity, spastic paralysis.
Porencephaly is characterized by the formation of cystic cavities within the cerebral parenchyma. The causes of the pathology are hemorrhage inside the brain, heart attacks, strokes. The typical location of the cyst is the Sylvian fissure.
In rare cases, porencephaly is characterized by the formation of abnormal passages between the cystic cavity and the ventricular spaces. The nosology is often characterized by other stigmas of disembryogenesis:
- Microcephaly;
- Encephalocele;
- Agiriya.
A child with a developmental defect experiences neurological disorders:
- Oligophrenia (mental retardation);
- Epilepsy attacks;
- Atrophic changes in the optic discs;
- Tetraparesis.
Concomitant changes complicate the clinical picture:
- Arterial and venous hemorrhages;
- Intracranial hypertension (in case of impaired circulation of cerebrospinal fluid);
- Cerebral infarctions;
- Hemiparesis;
- Focal epileptic foci.
Pseudoporencephalic cysts often have a unilateral (one-sided) location. They are often combined with defects of the central nervous system and defects in cell migration.
Schizencephaly on MRI
Holoprosencephaly - what is it?
The disease occurs due to the pathological division of the chemical substance prosencephalon.
Classification of nosology by severity:
- Lobarnaya;
- Semi-lobar;
- Alobar.
The heaviest is the last form. With it, a number of congenital anomalies are observed:
- Underdevelopment of the premaxillary bone;
- Sinophthalmos;
- Cebocephaly.
Against the background of multiple anomalies, parts of the face are difficult to distinguish. Associated neuronal migration defects:
- Synostosis (fusion of nerve ganglia);
- Presence of one cerebral ventricle.
The etiological mechanisms of hereditary defects have not been established. Holoprosencephaly is fatal in most cases, but the incomplete form can lead to disability.
Prader-Willi syndrome
This disease is determined by the same genetic mutation as for Angelman syndrome. The difference is that in this case, a violation of the hereditary material occurs on the father’s side. The karyotype is normal (46XX or 46XY). The prevalence (1 case per 12-15 thousand newborns) approximately coincides with the prevalence of Angelman syndrome.
Characteristic signs of Prader-Willi syndrome are the following symptoms:
· in the prenatal period, low fetal mobility;
· incorrect position of the fetus is common;
Possible hip dysplasia;
· by the age of two, a tendency to eat a lot (more than normal) may appear, which leads to obesity;
· low muscle tone (hypotonia), combined with impaired coordination of movements;
· feet and hands are usually small, and short stature is also characteristic;
· formation of strabismus and scoliosis;
· note increased drowsiness;
· bone density is at a lower level than in healthy people;
· saliva is thick, teeth are usually in poor condition;
· insufficient function of the gonads, ultimately causing infertility;
· later puberty compared to peers;
· patients learn to speak later and lag behind in mental development;
· external signs include a pronounced bridge of the nose, a narrow and high forehead, almond-shaped eyes, and narrow lips.
In most cases, a person with the mutation will have one to five signs of the disease.
Diagnosis of the disease is carried out by molecular genetic testing, for which children with reduced muscle tone are referred. Often, instead of a correct diagnosis, the more common “Down syndrome” is determined. An experienced geneticist who often encounters manifestations of Prader-Willi syndrome is able to diagnose it based on a set of external signs.
Symptoms of lissencephaly
Clinical manifestations of the nosology are determined immediately after the birth of the child. Not only genetic defects are noted, but also swallowing disorders in the baby, increased muscle tone, and developmental delay from peers.
Symptoms of lissencephaly in a child 2-5 months old:
- Oligophrenia;
- Mental development disorder;
- Myoclonus;
- Increased blood pressure.
At first, mental retardation is not evident. After the first year of life, disorders of psycho-speech skills are observed. Children are irritable, anxious, and muscle activity is reduced.
When compared with peers, there is a significant lag in development, formation of the muscular system, innervation of internal organs, and the pelvis.
With a mild course of the disease, the child can live until adolescence. External anomalies limit social communication. Patients rarely live to be eighteen years old, but there are about twenty types of pathology. Manifestations of the first type:
- Macrogyria;
- Schizencephaly;
- Microcephaly;
- Four-layer structure of the cortex;
- Pontine hypoplasia;
- Underdevelopment of the cerebellum.
It is difficult to determine lissencephaly in the absence of positive results of intrauterine ultrasound. The clinical picture appears after birth. Muscle paralysis and cramps appear immediately or during the first year.
In most cases, ultrasound examination will help establish the diagnosis of lissencephaly. A doctor may order magnetic resonance imaging for a pregnant woman and baby. MRI is harmless, but is not performed in the first trimester due to the lack of practical information about the effect of the magnetic field on the fetus.
Angelman syndrome
With Angelman syndrome, a characteristic set of pathological changes develops. In particular, there is a delay in psychological development, accompanied by problems with sleep, frequent chaotic movements (mostly with the hands), constant smiling and laughter.
Pathology develops in the absence of certain genes located on chromosome 15. In this case, a mandatory condition is the transfer of a mutant copy of the gene from the mother. If the damaged chromosome is inherited from the father, Prader-Willi syndrome will develop. The karyotype is usually normal (46XX and 46XY for girls and boys, respectively). Various independent studies indicate that the disease is associated with the UBE3A gene, which normally produces an enzyme component in a complex protein degradation system.
The frequency of occurrence of the syndrome is approximately 1 case per 10-20 thousand newborns (indicators differ among different scientists).
The characteristic features of patients with Angelman syndrome are the following:
· nutritional problems that begin during breastfeeding, as children do not gain weight well (prevalence of the symptom is about 75 percent);
· inhibited development of general motor skills, that is, children begin to sit and walk later than others;
· All children are characterized by speech development disorders;
· patients usually understand more than they are able to express using a limited vocabulary;
· often the disease is accompanied by attention deficit and hyperactivity;
· problems with learning in a regular school;
· 80% of patients develop epilepsy, accompanied by disturbances noticeable on electroencephalography; Scientists believe that epilepsy is a secondary (symptomatic) disease.
· performing unusual movements, which include random chaotic movements of the limbs, small tremors;
· occurrence of attacks of laughter in the absence of visible reasons;
· characteristic walking on stiff legs, which gave rise to comparisons with puppets;
· a head reduced in comparison with the average size, often with a flattened occiput;
· in some cases, there are peculiar memorable facial features - a wide mouth with sparsely spaced teeth, a protruding chin with a protruding tongue;
· various sleep disorders;
· in approximately 40 percent of cases, strabismus develops;
· about 10% of patients also suffer from spinal curvature;
· high temperatures are perceived with increased sensitivity;
· the greatest comfort is usually achieved in water (for example, in a bath)
As a rule, the syndrome is determined using molecular genetic diagnostic methods using chromosome 15. An indication for testing for a newborn is decreased muscle tone (hypotonia), a noticeable delay in the development of speech and fine motor skills. In addition, the disease may be indicated by small tremors, jerky, erratic movements, and walking on stiff legs.
The analysis can be carried out through fluorescence in situ hybridization, DNA methylation in the 15q11-q13 region. You can also check for mutations in the imprinting center and in the UBE3A gene.
Since the disease is caused by a genetic disorder, there is no adequate and effective treatment for it. Carrying out therapeutic measures, such as massage for patients with hypotension, can improve the quality of life.
The first signs of lissencephaly in newborns
Early detection of nosology helps to prolong the baby’s life, but it cannot save him from death. The first signs in newborns:
- Small head;
- Apathy, lethargy;
- Wide interocular distance;
- Epileptic convulsive syndrome;
- Hypertonicity of muscles;
- Increased width between the lungs and kidneys;
- Activation of pathological reflexes;
- Sloping frontal part.
During the first year of life, other manifestations appear - speech disorders, urinary incontinence, increased breathing and heart rate.
Lethal pterygum (Norman-Roberts) syndrome occurs in type 1 lissencephaly. A number of manifestations are added to the above-described manifestations:
- Difficulty sitting;
- Inability for the baby to maintain a vertical position;
- The appearance of generalized muscle twitching;
- Anomalies of the craniofacial region (wide eyes, sloping forehead, bumps on the back of the head);
- Ocular nystagmus;
- Decreased muscle tone.
Cerebellar ataxia is characterized by a lack of coordination. The child cannot maintain balance or maintain an upright position. Parents first pay attention to the difficulties of planting and maintaining a horizontal position.
Diagnosis of lissencephaly
The diagnosis is made immediately after birth using ultrasound, CT and MRI. Manifestations can be detected in utero from 20 weeks. Detection of the slightest disturbances in the development of the fetal brain requires additional diagnostics. Depending on the degree of damage, a decision is made on the possibility of terminating the pregnancy. If hereditary forms are suspected, genetic counseling is carried out for all family members.
Late verification of pathology requires constant conservative treatment. Therapy for severe forms is carried out in a hospital under the supervision of qualified doctors.
The main types of diagnosis of lissencephaly:
- Magnetic resonance imaging (MRI of the head) shows soft tissue. The study reveals cerebral anomalies, cysts, fluid accumulations, inflammatory foci;
- Intrauterine ultrasound of the head is performed at 22-27 weeks, when the process of furrow formation is observed;
- Computed tomography (CT) helps to verify changes in gray and white matter, additional solid formations. The examination leads to radiation exposure of tissues, so for children it is performed according to strict indications;
- Electroencephalography (EEG) verifies foci of increased brain activity and areas of parenchymal hyperactivity.
The most reliable study is MRI for lissencephaly in newborns, which allows you to determine the smallest anomalies.
Diagnosis and medical options
Typically, in this case, the diagnosis is made at birth or a little later, after an ultrasound scan and examination of the results of a CT or MRI.
But doctors may suspect the presence of pathology even during pregnancy, because they regularly perform ultrasounds of the fetus and constantly study its condition, in particular the development of the brain.
If the doctor suspects lissencephaly, in addition to ultrasound, several more diagnostic methods will need to be performed, for example, analysis for the clarity of mutated genes and nuclear magnetic resonance imaging.
This pathology can be detected no earlier than the 20th week of pregnancy, since the surface of the brain until this time, and without any disturbances, is smooth. It is after this period that deviations in fetal development can be noticed.
It is impossible to cure this defect; symptomatic treatment is possible, and it depends on the stage of development and location of the defects. Constant care must be maintained.
For people with hydrocephalus, shunt surgery is usually done, and special medications are taken to control seizures.