Больше информации про другие виды заболеваний на букву «Н»: Нарушение сна; Нарколепсия; Наследственная мозжечковая атаксия Пьера-Мари; Нарушения спинномозгового кровообращения; Невралгия тройничного нерва; Невралгия подчелюстного и подъязычного узлов; Невралгия языкоглоточного узла; Невралгия ушного узла; Неврастения; Невральная амиотрофия Шарко-Мари-Тута; Невринома слухового нерва; Невринома; Неврит зрительного нерва; Неврит глотки; Неврит лицевого нерва; Неврит; Невроз навязчивых состояний; Невроз глотки; Неврозы; Неврозоподобное заикание; Невропатия бедренного нерва.
Общее понятие
Невральная амиотрофия Шарко-Мари-Тута — хроническое заболевание организма, носящее наследственный тип образования. Характерным показателем является поражение нервной системы периферического отдела. Аномальные процессы проявляются в формировании и изменений структуры в мышечных зонах, например: уменьшение в размерах районов в начале нижних конечностей, а после — верхних. В комбинации с аномалией данного вида у больного может возникнуть гипестезия и снижение рефлекторной способности сухожилий, подергивания разных отделов мышц.
Врачи прибегают к различным вариантам обследования пациента с таким недугом, к ним относятся: электромиография, электронейрография, генетическое обследование, ДНК-проверка, биопсия нервов и мышечных участков. Радикальных приемов лечения — не существует, но имеются методики направленные на избавление человека от симптомов. Медики предписывают разнообразные витаминные комплексы, антихолинэстеразной способы, метаболические приемы, микроциркуляторные аналоги, ЛФК, физиотерапию и прочее.
Лечение болезни Шарко
Все способы лечения болезни Шарко-Мари-Тута не радикальны. Симптоматическое лечение включает медикаментозную терапию, физиотерапию, лечение у ортопеда и т.п.
Физиотерапевтическое лечение болезни Шарко-Мари-Тута включает ЛФК, массаж, электрофорез, диадинамотерапию, терапию лечебными грязями, разные виды ванн и др.
Медикаментозная терапия направлена на улучшение питания мышечных волокон. С этой целью назначают кокарбоксилазу, глюкозу, аденозинтрифосфат и др. Так же широко применяют антиоксидантные средства, препараты, улучшающие микроциркуляцию, и витамины. Хорошо зарекомендовали себя препараты, которые тормозят активность ацетилхолинэстеразы и повышают уровень ацетилхолина, например, прозерин, галантамин.
Дальнейшие разработки новых препаратов, направленные на радикальные меры – это мир без болезни Шарко-Мари-Тута. Прогрессирование болезни Шарко-Мари-Тута не отражается на том, сколько живут пациенты.
В Юсуповской больнице специалисты долгие годы помогают пациентам держать болезнь под контролем. Минимальная выраженность симптомов и медленное прогрессирование – результат работы врачей. В комфортных палатах, на новых тренажерах, в хорошо оснащенных кабинетах – вот где проходит лечение болезни Шарко-Мари-Тута. Не затягивайте с лечением, запишитесь на консультацию.
Полезные сведения
Невральная амиотрофия Шарко-Мари-Тута (ШМТ) входит в список хронических заболеваний (полиневропатий) генетического характера, которые проявляются в повышенном прогрессировании. К ней относятся:
- Синдром Русси-Леви.
- Гипертрофическая невропатия Дежерина-Сотта.
- Болезнь Рефсума и другие более редкие патологические процессы.
Согласно среднестатистическим данным ученые заявляют, что практически 83% случая вызваны в результате наследственной предрасположенности человека. Неблагоприятные действия в теле возникают по большей мере у мужчин, женщины менее подвержены описываемому заболеванию. Ссылаясь на многочисленные сведения, невральная амиотрофия ШМТ диагностируется с частотой от двух до тридцати шести случаев на сто тысяч населения планеты. Обычно, она носит семейный характер. Однако у членов одной семьи симптоматика клиника может быть по-разному выражена. Вместе с этим врачи наблюдают и спорадические варианты ШМТ. Медицинские сотрудники отмечают четкую взаимосвязь с потеряй координации по Фридрейху. У некоторых больных в разных ситуациях замечаются типичные симптомы одной или другой болезни. Иногда, с течением долгих лет, клиника одной аномалии может замениться симптомами другой.
Причины
Генетические заболевания определяются комбинацией генов для конкретного признака, которые находятся на хромосомах, полученных от отца и матери.
Человек получивший один нормальный и один ген заболевания, является носителем, но обычно не проявляет симптомов.
- Риск для двух родителей-носителей передачи дефектного гена детям – 25%.
- Иметь ребенка – носителя -50%.
- Шанс для ребенка получить нормальные гены – 25%.
Риск одинаковый для мужчин и женщин.
Доминантные генетические расстройства возникают, когда для появления болезни необходима только одна копия аномального гена. Аномальный ген может быть унаследован от любого из родителей или быть результатом новой мутации (изменения гена).
Риск передачи ненормального гена от пострадавшего родителя к потомству составляет 50% для каждой беременности независимо от пола ребенка.
Х-сцепленные доминантные генетические нарушения вызваны аномальным геном на Х-хромосоме. Мужчины с аномальным геном страдают более сильно, чем женщины.
Наследственная нейропатия подразделяется на несколько типов, называемых CMT1, CMT2, CMT3, CMT4 и CMTX.
CMT1
Является доминирующей формой расстройства, при котором скорости проводимости нерва являются медленными. Более распространен, чем CMT2. Вызван аномальными генами, которые участвуют в структуре и функции миелина. Дополнительно подразделен на CMT1A, CMT1B, CMT1C, CMT1D, CMT1X на основе специфических аномалий.
- CMT1A появляется из-за дублирования гена PMP22, который расположен на хромосоме 17 при 17p11.2. Является наиболее распространенным типом.
- CMT1B вызван аномалиями в гене MPZ на хромосоме 1 при 1q22.
- CMT1C появляется от аномалий SIMPLE, расположенном на 16 хромосоме при 16p13.1-p12.3.
- CMT1D аномалия EGR2, расположенной на 10, при 10q21.1-q22.1.
- CMT1X возникает от мутаций GJB1 (Xq13.1), Он кодирует белок связного соединения connexin32.
Узнать больше Что такое синдром Шихана?
CMT2
Является аутосомно-доминирующей формой расстройства, при котором скорости проводимости нерва обычно нормальны или немного медленнее, чем обычно. Вызван аномальными генами, участвующими в структуре и функции аксонов. Дополнительно подразделен на CMT2A-2L на основе мутаций.
- CMT2A, является наиболее распространенным и обусловлен ошибками MFN2, расположенным на хромосоме 1, в 1p36.2.
- CMT2B от мутаций RAB7 на хромосоме 3 при 3q21.
- CMT2C вызывается неизвестным геном на 12 – 12q23-34.
- CMT2D ошибки GARS, на 7 – 7p15.
- CMT2E от NEFL, расположенном на 8 – 8p21.
- CMT2F ошибки гена HSPB1.
- CMT2L мутации HSPB8.
Доминирующая промежуточная DI-CMT. Она названа так из-за «промежуточной» скорости проводимости, неопределенности относительно того, является ли нейропатия аксональной или демиелинизирующей. Известно, что доминантные мутации в DMN2 и YARS вызывают этот фенотип.
CMT3
Также называемый болезнью Дежерин-Соттас, у индивидуумов с этим расстройством обнаружена мутация в одном из генов, ответственных за CMT1A, CMT1B, CMT1D, CMT4.
CMT4
Аутосомно-рецессивная форма состояния. Он подразделяется на CMT4A, CMT4B1, CMT4B2, CMT4C, CMT4D, CMT4E, CMT4F.
- CMT4A вызван аномалиями GDAP1. Ген находится на хромосоме 8 при 8q13-q21.
- CMT4B1 – аномалия MTMR2 на 11 – 11q22.
- CMT4B2 от аномалий SBF2 / MTMR13, на 11 при 11p15.
- CMT4C ошибки KIAA1985, на хромосоме 5 – 5q32.
- CMT4D мутации NDRG1, на хромосоме 8 – 8q24.3.
- CMT4E, также известная как врожденная гипомиелиническая невропатия. Происходит от аномалий EGR2, на 10 – 10q21.1-q22.1.
- CMT4F аномалии PRX, на хромосоме 19 – 19q13.1-q13.2.
- CMT4H ошибки FDG4.
- CMT4J мутации FIG4.
Однако большинство случаев CMT2 не вызваны мутациями этих белков, поэтому многие генетические причины еще не обнаружены.
CMTX
Является X-связанной доминирующей формой расстройства. На CMT1X приходится около 90% случаев. Конкретный белок, ответственный за оставшиеся 10% CMTX, еще не идентифицирован.
Аутосомно-рецессивный CMT2 происходит из-за мутаций LMNA, GDAP1.
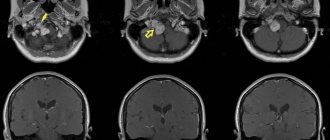
Структурное выравнивание с элементами вторичной структуры, расположенными сверху. Десять мутаций, вызывающих CMT, отмечены вертикальными стрелками. (Нажмите, чтобы увеличить)
Патогенез невральной амиотрофии Шарко-Мари-Тута
В медицине на данный момент не имеется точной и достоверной информации о происхождении и механизме возникновения невральной амиотрофии. Согласно проведенным экспериментам стало понятно только то, что почти у 75% пациентов с таким диагнозом, которые прошли генетическую консультацию, выделялось повторение определенной зоны 17-й хромосомы. Уже известно, что заболевание обладает несколькими группами, вызываемыми различными мутациями генов. Так, например, во время исследования больного с ШМТ, проявляющейся из-за мутации белка гена MFN2, начинается появление частиц митохондрий. В результате чего у человека происходит сбой в движении этих элементов по аксону.
Большое количество форм проявляются вследствие поражения миелинового наружного слоя волокон. Намного реже можно заметить формы с отклонением от нормы аксонов — объектов осевого направления, которые проходят в центральной части нервной структуры. Аномальные процесс способны повлиять на состояние передних и задних отростков спинного мозга, нейронов передних рогов, путей Голля и столбы Кларка. Повторно из-за проблем с функционированием периферического отдела, начинает образовываться мышечная недостаточность.
Разновидности и классификация
В неврологическом направлении медицины невральная амиотрофия Шарко-Мари-Тута разделена на две четких группы, которые клинически достаточно схожи между собой, но имеют список особенностей, позволяющих провести подобное разграничение:
- Невральная амиотрофия I типа — вызывает снижение скорости проведения нервного импульса.
- НА II типа — скорость подвергается пагубному влиянию в меньшей доли, проявляется отклонение нейрита.
Как диагностировать ШМТ?
Врач спросит о семейном анамнезе, и выявит признаки мышечной слабости — снижение мышечного тонуса, плоскостопие или высокий свод стоп (кавус).
Для исследования нервной проводимости проводится измерение силы и скорости электрических сигналов, которые проходят через нервы (Электромиография). Электроды помещаются на кожу, и вызывают слабые поражения электрическим током, которые стимулируют нервы. Задержанный или слабый ответ предполагает расстройство нервной системы, и, возможно, ШМТ.
При электромиографии (ЭМГ) тонкую иглу вводят в мышцы. Когда пациент расслабляет или сокращает мышцы, измеряется электрическая активность. Тестирование различных мышц покажет, какая из них страдает.
Генетическое тестирование проводится с помощью пробы крови, которая может показать, имеет ли пациент мутации гена.
В чем замечены проявления невральной амиотрофии Шарко-Мари-Тута?
При описываемом заболевании у человека первым делом берет начало формирование схожих мышечных некрозов в нижних конечностей. Первые проявления, как правило, происходят такое в возрасте двадцати лет (намного реже, врачи могут диагностировать симптоматику в подростковый период от 16 лет, а также до 30 лет). Подобные проявления состоят в основном в высокой степени утомляемости ног во время долгого нахождения в вертикальном положении. Замечен синдром так называемого «топтания», то есть для того, чтобы пациент мог снять дискомфортные ощущения, он начинает ходить на месте. В некоторых редких ситуациях НА проявляется расстройстве чувствительности тех же отделов, чаще всего — образуются парестезии в виде ползания мурашек.
Типичным ранних звоночком ШМ, считается полное отсутствие ахилловых и коленных рефлексов сухожилий. В итоге у больного проявляется свисание свода, неспособность ходить на пятках и анормальная походка, которая схожа с лошадиной. Впоследствии пагубный процесс увеличивает степень прогрессирования дальше и затрагивает мышцы и сгибатели. Максимальная уровень омертвения может привести к полной деформации ног с высоким сводом, схожу на тип стопы Фридрейха. Постепенно ШМТ переходит на проксимальные участки, к которым относятся голени и нижние зоны бедер. У пациента начинают серьезные деформации: свисающие стопы, ноги становятся нестандартной формы. Далее поражаются руки, кисть становится похожа на обезьянью лапу.
Стоит знать, что пагубные поражения никогда не поражают мышцы шейного периметра, туловища и плечевого района. Помимо вышеперечисленных симптомов, у больного могут появиться следующие проблемы:
- Слабое подергивание.
- Гипертрофия компенсаторной природы мышечных областей.
- Сенсорные нарушения.
- Возможность появления цианоза и отечности на кожном покрове.
Для ШМТ типично медленное развитие симптоматики. Период развития процесса уменьшения в размерах ног и рук может занять до десяти лет. Даже при таких серьёзных деформациях тела больной долгое время способен сохранять работоспособность и нормально выполнять различные бытовые обязанности. Ускорителями симптомов могут стать следующие факторы:
- Попадание в тело инфекционных агентов.
- Долгое пребывание на холоде.
- Травмирования головы.
- Повреждения спины и спинного мозга.
- Нехватка витаминов в организме.
Симптомы ШМТ у взрослых людей
- Слабость в мышцах ног и лодыжек;
- Искривление пальцев ног;
- Трудности подъема стопы из-за слабых мышц голеностопного сустава;
- Онемение в руках и ногах;
- Изменение формы голени, при этом нога становится очень тонкой ниже колена, в то время как бедра сохраняют нормальный объем мышц и форму (нога аиста);
- Со временем руки ослабевают и пациентам трудно выполнять повседневную работу;
- Появляются боли в мышцах и в суставах, человеку тяжело ходить. Нейропатическая боль возникает вследствие поврежденных нервов;
- В тяжелых случаях пациент может нуждаться в коляске, в то время как другие могут использовать специальная обувь или другие ортопедические устройства.
Варианты диагностики
Медицинские работники для диагностирования НА Шарко-Мари-Тута опираются на такие проявления, как:
- Возраст, в котором появились первые признаки недуга.
- Стандартная клиническая картина.
- Схожий характер разрушения организма.
- Медленная степень развития атрофий.
- Увеличение опасности симптомов.
Во время посещения кабинета специалиста, человеку проведут полноценный осмотр тела, чтобы выявить наличие мышечной слабости в стопах и голенях, изменения, отсутствие или серьезное снижение уровня рефлекторных способностей ахиллова и коленного отдела, гипестезию. Для того, чтобы отличить ШМТ от других нервно-мышечных расстройств врачи составляют ряд обследований, которые потребуется пройти инфицированному. К ним относятся:
- Электромиографическое исследование.
- Электронейрография.
- Анализ кровеносных телец на сахар, гормональную норму и наркотические вещества.
- Проведение консультации у специалиста в области генетики.
- ДНК-оценка или секвенирование генома (последний вариант очень дорогостоящий для широкого применения).
- Биопсия.
Симптомы
Заболевание Шарко Мари Тута может проявляться по-разному и главное вовремя узнать развитие болезни. Это нужно для того чтобы своевременно обратиться в медицинское учреждение.
Если пациента беспокоят некоторые признаки, то не рекомендуется игнорировать симптомы и посетить врача. У детей заболевание практически протекает незаметно, поэтому выявить патологию можно только у ребенка старше 14 лет.
Основные симптомы при развитии заболевания:
- Ноги начинают быстро уставать и пациенту проще шагать на месте, чем просто стоять.
- Заметно, как происходит снижение чувствительности нижних конечностей и по всему телу ощущаются мурашки.
- Если патология в запущенной форме, то может наблюдаться мышечная атрофия. Мышцы на ноге могут становится меньше в объёме. Если присмотреться на нижние конечности, то можно заметить, как они стали в виде перевернутой бутылке. Может происходить деформация ступней.
- Часто возникает неправильная походка и пациент может идти и поднимать высоко колени.
Невральная амиотрофия может развиваться незаметно и медленно. Если наследственная нейропатия Шарко–Мари–Тута прогрессирует уже пару лет, то у больного может быть повреждение нервных тканей, как рук, так и ног.
Перемежающийся дистрофический синдром возникает чаще всего в предплечье и кистях. Мышцы туловища поражаются и со временем происходит их атрофия. Тогда и возникает деформация позвоночника.
Спровоцировать ускорение патологии и ухудшить самочувствие могут инфекции, интоксикация и травмы. Если стали заметны даже небольшие подозрения на невральную амиотрофию, то необходимо не медлить и обращаться к врачу.