Hippel-Lindau disease (Cerebroretinal angiomatosis)
The onset of neurological manifestations usually occurs in the 3rd-4th decades of life. In childhood, Hippel-Lindau disease is characterized by the appearance of neurological symptoms against the background of pre-existing visual disorders. In some cases, the disease in children manifests itself with subarachnoid hemorrhage.
CNS damage
. The most common source of primary symptoms is cerebellar cysts (cerebellar cysts). They manifest with general cerebral symptoms (diffuse headaches, nausea not associated with food intake, vomiting, tinnitus) caused by increased intracranial pressure. The first signs also include epileptic seizures; they can be generalized or focal. Over time, signs of cerebellar damage appear, forming the symptom complex of cerebellar ataxia: static and dynamic discoordination, adiadochokinesis, hypermetria and asynergia, intention tremor, myodystonia. As the cerebellar tumor grows, displacement and compression of the brain stem occurs, accompanied by brain stem symptoms, primarily swallowing disorder, diplopia, and dysarthria. Spinal tumors (usually angioreticulomas) are manifested by radicular syndromes, loss of deep types of sensitivity, and absence of tendon reflexes. In 80% of cases of spinal pathology, a clinical picture similar to syringomyelia is observed. A picture of complete damage to the diameter of the spinal cord is possible.
Eye damage
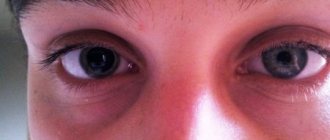
in the early stages they are diagnosed only by ophthalmoscopy. After 8 years, complaints about image hazyness and distortion (metamorphopsia) appear. In half of the patients, both eyes are affected. Retinal angiomas that increase over time lead to circulatory disorders in its vessels, ischemia and cystic degeneration. In the late stage, uveitis, cataracts, retinal detachment, glaucoma, and hemophthalmos are possible.
Kidney damage
in 60-90% of cases it is represented by cysts, in 45% of cases - renal cell carcinoma. As a rule, renal carcinoma clinically debuts between the ages of 40 and 50 years in patients who have previously been treated for neoplasms. In half of the cases, at the time of diagnosing carcinoma, its metastases are detected. The combination of polycystic kidney disease with retinal angiomatosis is more typical than its combination with cerebral angiomas. In 35% of patients with Hippel-Lindau disease, polycystic disease is diagnosed posthumously. In childhood, with a familial type of the disease, polycystic kidney disease is often its only manifestation.
Pheochromocytoma
in almost half of the cases it is bilateral. May be the only clinical manifestation of the disease. In combination with renal carcinoma it is quite rare.
Pancreatic damage
from 30 to 72% are cysts. Pancreatic cysts are benign and rarely lead to clinically significant pancreatic enzyme deficiency. Although there are known cases of complete replacement of normal gland tissue by a cyst with the development of diabetes mellitus.
Hippel-Lindau syndrome
Von Hippel-Lindau hereditary cancer syndrome (VHL) (cerebro-retino-visceral angiomatosis) is an autosomal dominant familial tumor syndrome that predisposes to the formation of various malignant and benign tumors. The most commonly observed with this syndrome are hemangioblastomas of the retina, cerebellum and spinal cord, renal cell carcinoma (RCC) of the kidneys, pheochromocytomas, and pancreatic tumors. In addition, cysts in the kidneys and pancreas, changes in the lungs and liver may occur. These tumors are characterized by high vascularity and overproduction of VEGF and other angiogenic proteins, as well as overproduction of erythropoietin, which can lead to increased red blood cell production and consequently polycythemia.
Mutations in the VHL lead to different types of the disease: type 1 is characterized by a low risk of developing pheochromocytoma, type 2 is characterized by a high risk of developing pheochromocytoma. In this case, type 2 is divided into three more subtypes: 2A is characterized by a low risk of developing RCC , 2B is characterized by a high risk of developing RCC, and with type 2C only pheochromocytoma develops without hemangioblastomas and RCC.
Hemangioblastomas develop in types 1, 2A and 2B. Mutations leading to the development of VHL type 1 syndrome are mainly microdeletions/insertions, nonsense mutations and large deletions (56% of cases). Missense mutations are responsible for 96% of type 2 cases, and mutations at codon 238 are responsible for the development of 43% of type 2 tumors.
VHL syndrome occurs in 1:35,000 newborns. The first symptoms appear in the second to fourth decades of life, and death occurs on average at 41 years of age. Half of the patients have only one sign of the disease. The diagnosis is made in a family case when one symptom appears, in the absence of a family history - if the patient has more than two hemangioblastomas or one hemangioblastoma and one visceral tumor. The VHL gene (OMIM 608537), mutations in which lead to the development of von Hippel-Lindau hereditary cancer syndrome (OMIM 193300), is a tumor suppressor gene. The gene is localized in the region of chromosome 3p25 and consists of 3 exons. The pVHL protein consists of 213 amino acid residues.
The Center for Molecular Genetics searches for mutations in the VHL using direct sequencing.
Large deletions occur in the VHL gene, affecting individual exons or the entire gene. To detect them, a method has been developed to determine the number of gene copies using quantitative MLPA analysis. To carry it out, you need fresh, unfrozen blood taken in a tube with EDTA.
When conducting prenatal (antenatal) DNA diagnostics in relation to a specific disease, it makes sense to diagnose common aneuploidies (Down, Edwards, Shereshevsky-Turner syndrome, etc.) using existing fetal material, paragraph 4.54.1. The relevance of this study is due to the high total frequency of aneuploidy - about 1 in 300 newborns, and the absence of the need for repeated sampling of fetal material.
Hippel-Lindau syndrome
The mechanism of pathology development
Hippel-Lindau disease is based on a generalized hereditary disorder, characterized by excessive growth of capillary tissue and the resulting appearance of a large number of tumor tumors.
The disease is transmitted to the next generation according to the autosomal dominant type, that is, if one of the parents has the gene encoding the pathology, the probability of the disease in the offspring is 50%.
The defect (mutation) leads to the suppression of a gene that suppresses tumor growth.
Oncological lesion of the pancreas
Oncological pathology of the pancreas is variable: tumors can be both benign and malignant. And in structure they are either cystic formations or neuroendocrine tumors.
Various oncological processes affecting the pancreas occur in half of patients with Hippel-Lindau disease. Symptoms are usually associated with dysfunction of the pancreas.
The average age of patients at the time of detection of tumors is 33-35 years. In the case of a malignant process, the metastatic process is directed to the liver.
In 1895, J. von Hippel [1] described a patient with a retinal angioma, and in 1926, A. Lindau [2] described a patient with a retinal angioma and hemangiomatosis of the central nervous system. A year later, the same author found an association of these manifestations with renal and pancreatic cysts [3]. The term von Hippel-Lindau syndrome (VHL) was coined by Melmon and Rosen [4]. This syndrome occurs in approximately 1 in 36,000 people [5] and is caused by a mutation in the 3p25/26 region, where the VHL tumor suppressor gene is localized [6–8]. 23% of patients have no family history of the disease [9–13].
The VHL gene was identified in 1993 [8, 14]. Approximately 20% of patients have a deletion of the VHL locus in the maternal or paternal allele [15, 16]. Germline VHL mutations are inherited in an autosomal dominant manner. Almost all VHL mutations in patients with pheochromocytoma are missense mutations. The VHL gene consists of 3 exons. The VHL protein (pVHL) contains 213 amino acid residues and has a molecular weight of approximately 28 kDa. Cells deficient in pVHL accumulate hypoxia-inducing factor (HIF), leading to overproduction of HIF-dependent products (which are involved in adaptation to hypoxia): vascular endothelial growth factor (VEGF), erythropoietin, and transforming growth factor alpha (TGF). This explains the strong vascularization of VHL-associated tumors [17–19]. Thus, the mutated VHL gene product leads to overregulation of various genes involved in the pathogenesis of hypoxia, accelerates angiogenesis, alters the extracellular matrix and cell cycle regulation [20–28]. However, the exact mechanisms of tumorigenesis in VHL syndrome currently remain unknown (Fig. 1).
Figure 1. VBC complex (VHL protein and Elongin B, C) and its mechanism of action. a — Normoxic condition — normal oxygen pressure; Ub—ubiquitin; HIF—hypoxia-inducing factor; pVHL—VHL protein; α - elongin C-binding domain of pVHL; β - β-domain (substrate binding domain) of pVHL; CUL2 - cullin-2, which forms a complex with elongin-B, C and pVHL; b — mutation of the VHL gene and, as a result, lack of regulation of HIF, VEGF, PDGF and TGF-α; c — mutation of the VHL gene and lack of regulation of aPKC λ (atypical protein kinase C) [29].
VHL syndrome is characterized by the development of retinal hemangioblastomas (retinal angioma) and central nervous system (CNS), bilateral and multifocal differentiated renal carcinoma, polycystic kidney disease, pheochromocytoma, cysts and neuroendocrine tumors of the pancreas, papillary cystadenoma of the epididymis in men and the broad ligament in women. , tumors of the inner ear. The damage to various organs and the degree of this damage are very variable (Tables 1, 2).
Clinically, the disease is divided into two groups. Type 1 involves primarily large deletions or mutations of the VHL gene and is characterized by a complete disease phenotype [retinal lesions, cysts or tumors of the brain and spinal cord, pancreatic, renal, and splenic cysts, solid pancreatic tumors (rarely adenocarcinomas), renal carcinomas, epididymal cystadenomas and tumor of the inner ear], but without pheochromocytoma. Type 2, in which pheochromocytoma develops (missense mutations of the VHL gene may also have an incomplete phenotype [13, 37, 40]. Type 2 is divided into subtypes with low (type 2A) and high (type 2B) risk of developing kidney cancer, as well as type 2C, manifested only by pheochromocytoma [35, 38, 41-43] (Table 3).
Hemangioblastomas of the central nervous system can be detected in childhood, but the average age of diagnosis is 29 years [32, 45, 46]. VHL-associated hemangioblastomas are detected on average 15 years earlier than sporadic ones [47]. Depending on the size and location of the tumor, clinical signs of hemangioblastoma of the central nervous system include headache, nausea, dizziness, ataxia, motor coordination disorder, nystagmus, and speech disorders. Hemangioblastoma of the spinal cord can lead to limb weakness and paresthesia. Diagnosis is made using MRI of the brain and spine. Hemangioblastomas are usually characterized by slow growth and have a high risk of bleeding, and are often multifocal. Understanding the pathogenesis of the disease is important for choosing the optimal timing of tumor screening and treatment [19]. The study of CNS tissues from deceased patients has helped to understand the histogenesis of hemangioblastomas [49]. Activation of hypoxia-inducing factor 2-alpha (HIF 2-α) occurs in small mesenchymal tumors and in the mesenchymal component of large tumors. Activation of HIF 1-α is observed in the epithelial component. This allowed us to assume that CNS damage in VHL syndrome is a long-term process of hemangioblastic proliferation and differentiation [50] (Fig. 2).
Figure 2. Hemangioblastoma of the central nervous system.
Ocular lesions are detected in approximately 37% of patients with VHL syndrome, of which only 14% have complete VHL deletion [51, 52]. Approximately 8% of patients have reduced visual acuity [53]. Laser or cryotherapy is used to treat retinal angioma [32, 34, 54]. Recent studies [55, 56] have shown that intravenous administration of an antagonist of vascular endothelial growth factor (anti-VEGF) for 7 months does not reduce the size of hemangioblastomas (Fig. 3).
Figure 3. Retinal angiomatosis.
Patients with VHL syndrome may experience both cysts and renal cancer [57–62]. The average age of manifestation is 37 years. CT and ultrasound are used for diagnosis [36, 63, 64]. Since solid cancers may contain cystic parts (which makes it difficult to differentiate between benign and malignant processes using imaging techniques), in the absence of evidence of metastases, treatment should be aimed at removing these formations, if possible, in compliance with the principle of organ-conserving surgery. This allows you to maintain renal function for as long as possible and avoid dialysis [58, 65]. Kidney tumors are characterized by slow growth (<0.5 cm/year) [66, 67]. The risk of metastasis correlates with tumor size. Surgical treatment is recommended for solid tumors >3 cm (US standards) or 5 cm (European standards) [58, 60, 67, 68]. Some authors [69] report a high risk of local recurrence (approximately 50%) at a mean time of 53 months (range, 10–115 months) and tumor growth at a rate of 34 mm/year (range, 1–10.8 mm). The gold standard for treating small tumors is open and laparoscopic partial nephrectomy. Currently, alternative methods are also used - cryotherapy and radiofrequency ablation [70]. The latter methods may affect the result of the pathological diagnosis, although, according to some data, the pathological diagnosis after the first cycle of cryotherapy in approximately 91% of cases confirms the results of the preliminary biopsy [71].
Pheochromocytoma is detected in approximately 26% of patients with VHL syndrome [37]. In patients with apparently sporadic pheochromocytoma, a VHL mutation is subsequently detected in 3–11% of cases [10, 12, 13]. Pheochromocytoma may be the first manifestation of the syndrome [30, 72]. In most cases, adrenal pheochromocytomas in VHL syndrome are bilateral (synchronous or metachronous) [37, 73]. Extra-adrenal pheochromocytomas occur in approximately 30% of cases [37, 74–76]. Pheochromocytomas, as part of the VHL syndrome, have an exclusively norepinephrine phenotype. Biochemical tumor markers can help distinguish VHL-associated pheochromocytomas from pheochromocytomas in MEN type 2 syndrome [75]. Differences in the biochemical phenotype between VHL syndrome and MEN type 2 are associated with different expression of tyrosine hydroxylase (TH), the rate-limiting enzyme in catecholamine synthesis, and phenylethanolamine-N-methyltransferase (PNMT). Patients with VHL syndrome have low expression of PNMT, which converts norepinephrine to epinephrine. Differences in biochemical phenotype are also associated with differences in catecholamine storage, transport and secretion [77]. MEN 2-associated pheochromocytomas contain higher concentrations of catecholamines due to greater TH expression. VHL-associated pheochromocytomas secrete catecholamines continuously, whereas in MEN 2 the secretion is episodic. This also determines the differences in the clinical manifestations of the two syndromes. For example, patients with MEN 2 more often complain of crisis increases in blood pressure [78]. In addition to genetic differences [26], different expression of erythropoietin and its receptor has been recorded [79]. Additionally, approximately 80% of pheochromocytomas are asymptomatic and are discovered incidentally on imaging studies. The low sensitivity of some radionuclide imaging techniques may be due to a relative paucity of storage granules or reduced expression of membrane norepinephrine or vesicular monoamine transporters [80]. Therefore, 123I-MIBG (metaiodobenzylguanidine) scintigraphy often fails to detect VHL-related adrenal pheochromocytomas [81, 82]. PET with 6-18F-fluorodopamine is a more sensitive method [36, 83]. Malignant pheochromocytomas with metastases in the lungs, liver, bones, and lymph nodes are rarely found in VHL syndrome [37, 74, 84—87]. Metastases are detected in less than 7% of cases [37]. Unfortunately, at present there are no clear signs that can reliably distinguish benign from malignant pheochromocytoma, although it is already known that germline mutation of the SDHB gene is an accurate marker in this regard [86–88]. Detection of pheochromocytoma in patients with VHL syndrome is especially important, given the high likelihood of surgical interventions for other tumors (hemangioblastomas of the central nervous system, etc.). Undiagnosed pheochromocytoma with other interventions can lead to life-threatening hypertensive crises. More than 70% of pheochromocytomas in children are VHL-associated. Every patient with VHL syndrome and confirmed pheochromocytoma should undergo 6-18F-fluorodopamine PET or 123I-MIBG scintigraphy to identify extra-adrenal pheochromocytoma or metastases before surgery [89]. Treatment of pheochromocytoma is surgical. At the same time, 6-month therapy with tyrosine kinase inhibitors leads to a tumor reduction by 21% and a reduction in plasma levels of normetanephrines and chromogranin A [90] (Fig. 4).
Figure 4. Bilateral pheochromocytoma and polycystic kidney disease.
Epididymal cystadenomas are benign tumors that can be bilateral [48]. More often they are about 2 cm in diameter and can spread to the spermatic cord, leading to infertility. Surgical treatment is usually not performed; observation is necessary [92].
35–75% of patients with VHL syndrome have benign cysts and microcystic adenomas of the pancreas [93–96]. According to CT data, pancreatic neuroendocrine tumors are detected in 17% of patients [95, 97, 98]. Of 633 patients with VHL syndrome, 108 (17%) had pancreatic endocrine tumors, and 9 (8%) had metastases [99]. Metastasis is more likely if the tumor size exceeds 3 cm. In 78% of patients with metastases (7 of 9), the mutation was localized in exon 3 of the VHL gene, and tumor doubling occurred in an average of 337 days. Such patients require surgical treatment. If the tumor size is less than 3 cm, it grows slowly, and the mutation is not localized in exon 3, then we can limit ourselves to monitoring the patient. The average age of diagnosis of pancreatic neuroendocrine tumors is 35 years. Pancreatic cysts occur in patients over 15 years of age and are often asymptomatic. Depending on the size and location, clinical symptoms may be caused by biliary obstruction and/or enzyme deficiency. In such cases, a biliary stent is installed and/or enzyme preparations are prescribed.
Hormone-active neuroendocrine tumors are rare [95, 100]. According to recent studies, the mortality rate for pancreatic endocrine tumors is 6%. In 60% of cases, scintigraphy detects somatostatin receptors; malignant tumors were detected in 58% of cases [101].
Tumors of the inner ear are located in the labyrinth, under the dura mater on the posterior surface of the pyramid of the temporal bone [102-104]. These tumors practically do not metastasize [105]. Symptoms include hearing loss, tinnitus, dizziness and/or facial palsy [106, 107]. Thus, in patients with a VHL mutation, an audiological examination and high-resolution CT or MRI of the inner ear are absolutely recommended, as early surgical intervention can preserve hearing. In patients with bilateral tumors causing deafness, hearing can be restored with a cochlear implant [108].
By the age of 60, 90% of VHL mutation carriers have some clinical manifestations of the syndrome [45]. Long-term prognosis and mortality are usually influenced by the presence of retinal and central nervous system hemangioblastomas, as well as advanced renal carcinoma [31–33]. Thus, timely examination and identification of pathology associated with VHL syndrome is the key to successful treatment and increasing the patient’s life expectancy (Table 4).
Hemangioblastoma of the central nervous system
The most commonly reported manifestation of von Hippel-Lindau syndrome is the development of hemangioblastoma of the cerebellum, spinal cord, or retina. A tumor developed on the cerebellum has the following manifestations:
- Headaches of a bursting nature.
- Nausea, vomiting.
- Uncertainty when walking due to poor coordination.
- Dizziness.
- Impaired consciousness (typical of later stages of pathology).
When hemangioblastoma is localized in the spinal cord, the main clinical symptoms are decreased sensitivity, paresis and paralysis, disturbances in the processes of urination and defecation. Pain syndrome is observed only occasionally.
This type of tumor is classified as slowly progressing. And the most informative examination method for diagnosis and control is considered to be magnetic resonance imaging (MRI), enhanced by contrasting the area under study.
The only effective treatment for cerebellar hemangioblastoma today is its surgical removal. The use of radiation and drug methods did not show convincing positive results.
After microsurgical removal, hemangioblastoma, as a rule, does not recur, but other neoplasms may appear. When surgically treating this tumor, it is necessary to take into account its multiplicity, which is typical for Hippel-Lindau disease.
Disease prevention
Hippel-Lindau disease traumatizes people not only physically, but also morally. The diagnosis usually shocks patients. This disease puts a person and his loved ones in an emergency situation. Fear of the onset of the disease, fear of waiting for the tumor to grow, fear of treatment - all this keeps the patient in almost constant tension.
There is no need to despair if this diagnosis is made. Although there is no specific advice to avoid the disease, regular examinations will help detect emerging tumors at an early stage. As long as they do not cause any symptoms, simple observation is usually sufficient. Further surgery may help avoid complications.
It is recommended to be examined by a specialist annually.
The development process is rapid and irreversible
There are several stages of development of this disease:
- Preclinical . It is characterized by initial accumulations of capillaries and their slight expansion.
- Classic . At this stage, typical retinal angiomas are formed.
- Exudative . It is characterized by high permeability of the walls of angiomatous nodes.
- At the fourth stage , retinal detachment occurs, and it can be tractional or exudative in nature.
- Terminal . At this stage, glaucoma, uveitis, and phthisis of the eyeball develop.
If you start treating Hippele Lindau disease at an early stage, you can achieve fairly effective results. In addition, thanks to timely therapy, it is possible to minimize the risk of complications.
Hippel-Lindau disease is characterized by intention tremor. What other diseases accompany this symptom?
Brain astrocytoma is a serious tumor that can be fatal. What methods of removing formations exist?
Where do tumors form?
As a rule, tumors affect the central nervous system, kidneys, eyes, adrenal glands, and pancreas. Tumors of the inner ear, appendages, and broad ligaments of the uterus are less common.
Cerebellar tumors (hemangioblastomas) cause severe headaches, nausea, or gait disturbances. Masses in the spinal cord cause numbness in limited areas of the skin and weakness in certain muscle groups. Retinal tumors (angiomas) can cause retinal detachment, affect vision, and even lead to blindness.
Tumors in the kidneys and pancreas are carcinomas, and therefore are mostly malignant. Symptoms make themselves felt too late and are already accompanied by metastases. Adrenal tumors can, through a large release of stress hormones, lead to headaches, rapid heartbeat, and sweating.
The table below shows the organs that may be affected by this disease, indicating the common and rare manifestations of Hippel-Lindau disease in percentage terms.
| Organ | Manifestation of the disease | Percent |
| 1. Eyes | Retinal angioma (hemangioblastoma) | 48% |
| 2. Central nervous system | Hemangioblastoma Astrocytoma | 60-80% 0,3% |
| 3. Kidneys | Kidney cancer Renal cyst | 25-45% 33% |
| 4. Pancreas | Numerous cysts Serous cystadenoma Pancreatic islet cell tumor | 17-56% 1% 8-17% |
| 5. Adrenal gland | Pheochromocytoma | 20% |
| 6. Ear | Tumor of the endolymphatic sac | 5-10% |
| 7. Adnexa and broad ligaments | Cystadenoma and cyst (in men) | 50% |
| 8. Pituitary gland | Adenoma | 0,3 % |
| 9. Liver | Cyst | 1% |
| 10. Spleen | Cyst | 0,3% |
Standard Treatments
There is no universal treatment recommendation. Treatment options can only be determined by careful assessment of the individual patient's overall situation—symptoms, test results, imaging studies, and general physical condition. The following are suggested as general recommendations for possible treatment options.
— Hemangioblastomas of the brain and spinal cord.
Symptoms associated with hemangioblastomas of the brain and spinal cord depend on the location, size of the tumor, and the presence of associated swelling or cysts. Symptomatic lesions grow faster than asymptomatic lesions. Cysts often cause more symptoms than the tumor itself. Once the lesion is removed, the cyst will collapse. If any part of the tumor remains in place, the cyst will fill again. Small hemangioblastomas that are asymptomatic and not associated with a cyst are sometimes treated with stereotactic radiosurgery, but this is more prevention than cure, and long-term results seem to show only modest benefit. Additionally, symptoms may not improve during the recovery period.
— Neuroendocrine tumors of the pancreas.
Careful analysis is required to differentiate serous cystadenomas from pancreatic neuroendocrine tumors (NETs of the pancreas). Cysts and cystadenomas usually do not require treatment. Pancreatic NETs should be assessed by size, behavior, and specific genetic mutation.
— Renal cell carcinoma.
Hippel-Lindau disease kidney tumors are often discovered when they are very small in size and in very early stages of development. The strategy for ensuring that a person has a sufficiently functioning kidney throughout their life begins with careful monitoring and choosing to operate only when the size of the tumor or high growth rate suggests that the tumor has metastatic potential (approximately 3 cm). In this case, the technique of organ-saving surgery is widely used. Radiofrequency ablation (RFA) or cryosurgery (cryotherapy) may be considered, especially for small, early-stage tumors. Care must be taken not to damage adjacent structures and to limit the formation of scars that may complicate subsequent operations.
— Retinal hemangioblastomas.
Small peripheral lesions can be successfully treated with minimal or no vision loss using laser. Large lesions often require cryotherapy. If the hemangioblastoma is located on the optic nerve head, there are several treatment options that will successfully preserve vision.
— Pheochromocytomas.
Surgical removal is performed after adequate blockade with medications; laparoscopic partial adrenalectomy is preferred. Vital signs are closely monitored for at least a week after surgery while the body adjusts to its “new normal.” Particular care is required during any type of surgery, as well as during pregnancy and childbirth. Even pheochromocytomas that do not appear active or cause symptoms should be considered for removal, ideally before pregnancy or non-emergency surgery.
— Tumors of the endolymphatic sac.
Patients whose tumor or hemorrhage is visible on MRI but who can still hear require surgery to prevent their condition from worsening. Deaf patients with tumor imaging findings should undergo surgery if other neurologic symptoms are present to prevent deterioration of balance. Not all endolymphatic sac tumors are visible on imaging; some are only discovered during surgery.