Болезнь Гиппеля-Линдау (Цереброретинальный ангиоматоз)
Дебют неврологических проявлений обычно приходится на 3-4-е десятилетия жизни. В детском возрасте болезнь Гиппеля-Линдау отличается появлением неврологической симптоматики на фоне уже существующих зрительных расстройств. В ряде случаев заболевание у детей манифестирует субарахноидальным кровоизлиянием.
Поражение ЦНС
. Наиболее часто источником первичных симптомов выступают церебеллярные кисты (кисты мозжечка). Они манифестируют общемозговыми симптомами (диффузными головными болями, тошнотой без связи с приемом пищи, рвотой, шумом в ушах), обусловленными повышением внутричерепного давления. К первым признакам также относятся эпиприступы, они могут быть генерализованными либо фокальными. Со временем проявляются признаки поражения мозжечка, формирующие симптомокомплекс мозжечковой атаксии: статическая и динамическая дискоординация, адиадохокинез, гиперметрия и асинергия, интенционный тремор, миодистония. По мере роста церебеллярного новообразования возникает смещение и сдавление мозгового ствола, сопровождающееся стволовыми симптомами, в первую очередь, расстройством глотания, диплопией, дизартрией. Спинальные опухоли (чаще ангиоретикуломы) проявляются корешковыми синдромами, выпадением глубоких видов чувствительности, отсутствием сухожильных рефлексов. В 80% случаев спинальной патологии отмечается клиника, сходная с сирингомиелией. Возможна картина полного поражения поперечника спинного мозга.
Поражение глаз
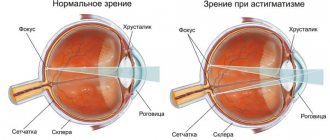
на ранних стадиях диагностируются лишь при офтальмоскопии. После 8 лет появляются жалобы на туманность изображения и его искажение (метаморфопсии). У половины пациентов выявляется поражение обоих глаз. Увеличивающиеся со временем ангиомы сетчатки приводят к расстройству кровообращения в ее сосудах, ишемии и кистозной дегенерации. В поздней стадии возможны увеит, катаракта, отслойка сетчатки, глаукома, гемофтальм.
Поражение почек
в 60-90% случаев представлено кистами, в 45% случаев — ренальноклеточной карциномой. Как правило, почечная карцинома клинически дебютирует в возрасте от 40 до 50 лет у больных, которые ранее уже лечились по поводу новообразований. В половине случаев на момент диагностирования карциномы выявляются ее метастазы. Сочетание поликистоза почек с ангиоматозом сетчатки более характерно, чем его комбинация с церебральными ангиомами. У 35% пациентов, имеющих болезнь Гиппеля-Линдау, поликистоз диагностируется посмертно. В детском возрасте при семейном типе заболевания поликистоз почек зачастую является его единственным проявлением.
Феохромоцитома
почти в половине случаев имеет двусторонний характер. Может выступать единственным клиническим проявлением болезни. В сочетании с почечной карциномой наблюдается довольно редко.
Поражение поджелудочной железы
от 30 до 72% составляют ее кисты. Кисты поджелудочной железы носят доброкачественный характер и редко приводят к клинически значимой ферментативной недостаточности панкреас. Хотя известны случаи полного замещения кистой нормальных тканей железы с развитием сахарного диабета.
Гиппеля-Линдау синдром
ВVon Hippel-Lindau hereditary cancer syndrome (VHL) (церебро-ретино-висцеральный ангиоматоз)– аутосомно-доминантный семейный опухолевый синдром, предрасполагающий к образованию различных злокачественных и доброкачественных опухолей. Наиболее часто при данном синдроме наблюдаются гемангиобластомы сетчатки, мозжечка и спинного мозга, renal cell carcinoma (RCC) почек, феохромоцитомы, опухоли поджелудочной железы. Кроме того, могут встречаться цисты в почках и поджелудочной железе, изменения в легких и печени. Эти опухоли характеризуются высокой васкуляризацией и избыточной продукцией VEGF и других ангиогенных белков, а также избыточной продукцией эритропоэтина, что может привести к увеличению продукции эритроцитов и, как следствие, к полицитемии.
Мутации в гене VHL приводят к разным типам заболевания: тип 1 характеризуется низким риском развития феохромоцитомы, тип 2 – высоким риском развития феохромоцитомы. При этом тип 2 делится еще на три подтипа: 2А характеризуется низким риском развития RCC, 2В – высоким риском развития RCC, а при типе 2С развивается только феохромоцитома без гемангиобластом и RCC.
Гемангиобластомы развиваются при типах 1, 2А и 2В. Мутации, приводящие к развитию синдрома VHL типа 1, представляют собой в основном микроделеции/вставки, нонсенс-мутации и крупные делеции (56% случаев). К 96% случаям типа 2 приводят миссенс-мутации, а мутации в кодоне 238 ответственны за развитие 43% опухолей при типе 2.
Синдром VHL встречается с частотой 1:35 000 новорожденных. Первые симптомы появляются на втором-четвертом десятилетии жизни, смерть наступает в среднем в 41 год. У половины больных встречается только один признак заболевания. Диагноз ставится в семейном случае при появлении одного признака, при отсутствии семейной истории – в случае наличия у пациента более двух гемангиобластом или одной гемангиобластомы и одной висцеральной опухоли. Ген VHL (OMIM 608537), мутации в котором ведут к развитию von Hippel-Lindau hereditary cancer syndrome (OMIM 193300) является геном-супрессором опухолевого роста. Ген локализован в области хромосомы 3р25 и состоит из 3 экзонов. Белок pVHL состоит из 213 аминокислотных остатков.
В Центре Молекулярной Генетики проводится поиск мутаций в гене VHL методом прямого секвенирования.
В гене VHL встречаются крупные делеции, затрагивающие отдельные экзоны или весь ген. Для их детекции разработан метод определения количества копий гена с помощью количественного MLPA-анализа. Для его проведения необходима свежая незамороженная кровь, взятая в пробирку с ЭДТА
При проведении пренатальной (дородовой) ДНК-диагностики в отношении конкретного заболевания, имеет смысл на уже имеющемся плодном материале провести диагностику частых анеуплоидий (синдромы Дауна, Эдвардса, Шерешевского-Тернера и др), пункт 4.54.1. Актуальность данного исследования обусловлена высокой суммарной частотой анеуплоидий — около 1 на 300 новорожденных, и отсутствием необходимости повторного забора плодного материала.
Гиппеля-Линдау синдром
Механизм развития патологии
В основе болезни Гиппеля-Линдау лежит генерализованное нарушение наследственного характера, характеризующееся избыточным разрастанием ткани капилляров и появлением в результате большого количества опухолевых новообразований.
Заболевание передается следующему поколению по аутосомно-доминантному типу, то есть при наличии гена, кодирующего патологию, у одного из родителей, вероятность заболеваемости потомка составляет 50 %.
Дефект (мутация) приводит к подавлению гена, являющегося супрессором опухолевого роста.
Онкологическое поражение поджелудочной железы
Онкологическая патология поджелудочной железы вариабельна: опухоли могут носить как доброкачественный, так и злокачественный характер. А по структуре это либо кистозные образования, либо нейроэндокринные опухоли.
Различные онкологические процессы, поражающие поджелудочную железу, встречаются у половины больных с болезнью Гиппеля-Линдау. Симптомы при этом связаны, как правило, с нарушениями функции поджелудочной железы.
Средний возраст пациентов на момент обнаружения новообразований 33-35 лет. В случае злокачественного процесса метастатический процесс направлен в печень.
В 1895 г. Ю. фон Гиппель [1] описал пациента с ретинальной ангиомой, а в 1926 г. А. Линдау [2] — пациента с ретинальной ангиомой и гемангиоматозом центральной нервной системы. Год спустя тот же автор обнаружил ассоциацию этих проявлений с почечными и панкреатическими кистами [3]. Термин «синдром von Hippel—Lindau» (VHL) был введен Мелмоном и Роузеном [4]. Этот синдром выявляется приблизительно у 1 из 36 000 человек [5] и обусловлен мутацией в участке 3p25/26, где локализован ген подавления роста опухоли VHL [6—8]. 23% пациентов не имеют семейного анамнеза заболевания [9—13].
Ген VHL был идентифицирован в 1993 г. [8, 14]. Приблизительно у 20% пациентов выявляется делеция VHL-локуса в материнской или отцовской аллели [15, 16]. Герминальные мутации VHL наследуются по аутосомно-доминантному типу. Почти все мутации VHL у пациентов с феохромоцитомой являются миссенс-мутациями. Ген VHL состоит из 3 экзонов. Белок VHL (pVHL) включает 213 аминокислотных остатков, и его молекулярная масса равна приблизительно 28 кДa. Клетки с дефицитом pVHL накапливают фактор, индуцирующий гипоксию (HIF), что приводит к перепроизводству HIF-зависимых продуктов (которые вовлечены в адаптацию к гипоксии): сосудистого эндотелиального фактора роста (VEGF), эритропоэтина и трансформирующего ростового фактора альфа (TGF). Это объясняет сильную васкуляризацию VHL-ассоциированных опухолей [17—19]. Таким образом, продукт мутированного гена VHL приводит к сверхрегулированию различных генов, участвующих в патогенезе гипоксии, ускоряет ангиогенез, изменяет внеклеточный матрикс и регуляцию клеточного цикла [20—28]. Однако точные механизмы туморогенеза при синдроме VHL в настоящее время остаются неизвестными (рис. 1).
Рисунок 1. Комплекс VBC (VHL protein and Elongin B, C) и механизм его действия. а — Normoxic condition — нормальное давление кислорода; Ub — убиквитин; HIF — фактор, индуцирующий гипоксию; pVHL — VHL-протеин; α — элонгин C-связывающий домен pVHL; β — β-домен (субстратсвязывающий домен) pVHL; CUL2 — куллин-2, формирующий комплекс с элонгином-B, C и pVHL; б — мутация гена VHL и, как результат, отсутствие регуляции HIF, VEGF, PDGF и TGF-α; в — мутация гена VHL и отсутствие регуляции aPKC λ (атипичная протеинкиназа C) [29].
VHL-синдром характеризуется развитием гемангиобластом сетчатки глаза (ангиомы сетчатки) и центральной нервной системы (ЦНС), билатеральной и мультифокальной дифференцированной карциномы почки, поликистоза почек, феохромоцитомы, кист и нейроэндокринных опухолей поджелудочной железы, папиллярной цистаденомы придатка яичка у мужчин и широкой связки у женщин, опухолей внутреннего уха. Поражение различных органов и степень этого поражения очень вариабельны (табл. 1, 2).
Клинически заболевание делится на две группы. Тип 1 включает главным образом большие делеции или мутации гена VHL и характеризуется полным фенотипом заболевания [поражение сетчатки, кисты или опухоли головного и спинного мозга, панкреатические, почечные, и селезеночные кисты, солидные панкреатические опухоли (реже аденокарциномы), карциномы почек, цистаденомы эпидидимуса и опухоль внутреннего уха], но без феохромоцитомы. Тип 2, при котором развивается феохромоцитома (миссенс-мутации гена VHL может иметь и неполный фенотип [13, 37, 40]. Тип 2 подразделяется на подтипы с низким (тип 2A) и высоким (тип 2B) риском развития рака почки, а также тип 2C, проявляющийся только феохромоцитомой [35, 38, 41—43] (табл. 3).
Гемангиобластомы ЦНС могут выявляться в детском возрасте, однако средний возраст диагностирования составляет 29 лет [32, 45, 46]. VHL-ассоциированные гемангиобластомы выявляются в среднем на 15 лет раньше, чем спорадические [47]. В зависимости от размера и местоположения опухоли клинические признаки гемангиобластомы ЦНС включают головную боль, тошноту, головокружение, атаксию, расстройство координации движений, нистагм, расстройства речи. Гемангиобластома спинного мозга может приводить к слабости конечностей и парестезиям. Диагноз устанавливается с помощью МРТ головного мозга и позвоночника. Гемангиобластомы обычно характеризуются медленным ростом и имеют высокий риск кровотечений, часто являются мультифокальными. Понимание патогенеза заболевания важно для выбора оптимального времени скрининга на опухоли и лечение [19]. Исследование тканей ЦНС умерших пациентов помогло пониманию гистогенеза гемангиобластом [49]. Активация фактора, индуцирующего гипоксию 2-альфа (HIF 2-α) происходит в маленьких мезенхимальных опухолях и в мезенхимальном компоненте больших опухолей. Активация HIF 1-α наблюдается в эпителиальном компоненте. Это позволило предполагать, что поражение ЦНС при VHL-синдроме — длительный процесс гемангиобластической пролиферации и дифференцировки [50] (рис. 2).
Рисунок 2. Гемангиобластома ЦНС.
Поражения глаз выявляются примерно у 37% пациентов с VHL-синдромом, среди них только у 14% обнаруживается полная делеция VHL [51, 52]. Приблизительно у 8% пациентов снижена острота зрения [53]. Для лечения ангиомы сетчатки используют лазерную или криотерапию [32, 34, 54]. Недавние исследования [ 55, 56] показали, что при внутривенном введении антагониста сосудистого эндотелиального фактора роста (anti-VEGF) в течение 7 мес размер гемангиобластом не уменьшается (рис. 3).
Рисунок 3. Ангиоматоз сетчатки.
У пациентов с синдромом VHL могут встречаться как кисты, так и рак почек [57—62]. Средний возраст манифестации — 37 лет. Для диагностики используют КТ и УЗИ [36, 63, 64]. Поскольку со`лидные раки могут содержать кистозные части (что затрудняет дифференцирование доброкачественных и злокачественных процессов с помощью визуализирующих методик), при отсутствии данных о метастазах лечение должно быть направлено на удаление этих образований по возможности с соблюдением принципа органосохраняющей операции. Это позволяет поддерживать почечную функцию максимально долго и избежать диализа [58, 65]. Опухоли почек отличаются медленным ростом (<0,5 см/год) [66, 67]. Риск метастазирования коррелирует с размером опухоли. Хирургическое лечение рекомендуется при размере со`лидных опухолей >3 см (по стандартам США) или 5 см (по стандартам Европы) [58, 60, 67, 68]. Некоторые авторы [69] сообщают о высоком риске местного рецидива (приблизительно 50%) в среднем в течение 53 мес (диапазон 10—115 мес) и росте опухоли со скоростью 34 мм/год (диапазон 1—10,8 мм). «Золотым» стандартом лечения небольших опухолей является открытая и лапароскопическая частичная нефрэктомия. В настоящее время используются также альтернативные методы — криотерапия и радиочастотная аблация [70]. Последние методы могут повлиять на результат патоморфологического диагноза, хотя, по некоторым данным, патоморфологический диагноз после первого цикла криотерапии приблизительно в 91% случаев подтверждает результаты предварительной биопсии [71].
Феохромоцитома выявляется примерно у 26% пациентов с синдромом VHL [37]. У пациентов с очевидно спорадической феохромоцитомой в 3—11% случаев впоследствии выявляют мутацию VHL [10, 12, 13]. Феохромоцитома может быть первым проявлением синдрома [30, 72]. В большинстве случаев надпочечниковые феохромоцитомы при VHL-синдроме двусторонние (синхронные или метахронные) [37, 73]. Вненадпочечниковые феохромоцитомы встречаются примерно в 30% случаев [37, 74—76]. Феохромоцитомы как часть синдрома VHL имеют исключительно норадреналиновый фенотип. Биохимические маркеры опухоли могут помочь отличить VHL-ассоциированные феохромоцитомы от феохромоцитом при синдроме МЭН 2-го типа [75]. Различия в биохимическом фенотипе при VHL-синдроме и МЭН 2-го типа связаны с различной экспрессией тирозингидроксилазы (TH) — лимитирующего фермента синтеза катехоламинов, и фенилэтаноламин-N-метилтрансферазы (PNMT). У пациентов с синдромом VHL отмечена низкая экспрессия PNMT, преобразующей норадреналин в адреналин. Различия биохимического фенотипа также связаны с различиями хранения, транспорта и секреции катехоламинов [77]. МЭН 2-ассоциированные феохромоцитомы содержат более высокие концентрации катехоламинов из-за более выраженной экспрессии TH. VHL-ассоциированные феохромоцитомы, секретируют катехоламины непрерывно, тогда как при синдроме МЭН 2 отмечен эпизодический характер секреции. Это определяет и различия клинических проявлений двух синдромов. Например, пациенты с МЭН 2 чаще жалуются на кризовые подъемы АД [78]. Помимо генетических различий [26], регистрируется разная экспрессия эритропоэтина и его рецептора [79]. Кроме того, около 80% феохромоцитом бессимптомны и выявляются случайно при визуализирующих исследованиях. Низкая чувствительность некоторых радионуклидных методов визуализации может объясняться относительной нехваткой гранул хранения или уменьшенной экспрессией мембранного норадреналина или везикулярных моноаминных транспортеров [80]. Поэтому сцинтиграфия с 123I-MIBG (метайодбензилгуанидином) часто не обнаруживает VHL-связанные надпочечниковые феохромоцитомы [81, 82]. ПЭТ с 6-18F-фтордопамином более чувствительный метод [36, 83]. Злокачественные феохромоцитомы с метастазами в легких, печени, костях, лимфоузлах редко встречаются при синдроме VHL [37, 74, 84—87]. Метастазы выявляются менее чем в 7% случаев [37]. К сожалению, в настоящее время нет четких признаков, позволяющих надежно отличить доброкачественную от злокачественной феохромоцитомы, хотя уже известно, что герминальная мутация гена SDHB, является в этом отношении точным маркером [86—88]. Выявление феохромоцитомы у пациентов с синдромом VHL особенно важно, учитывая высокую вероятность хирургических вмешательств по поводу других опухолей (гемангиобластом ЦНС и др.). Невыявленная феохромоцитома при других вмешательствах может привести к опасным для жизни гипертоническим кризам. Более 70% феохромоцитом у детей являются VHL-ассоциированными. Каждому пациенту с VHL-синдромом и подтвержденной феохромоцитомой до оперативного лечения необходимо проводить ПЭТ с 6-18F-фтордопамином или сцинтиграфию с 123I-MIBG для выявления вненадпочечниковой феохромоцитомы или метастазов [89]. Лечение феохромоцитомы оперативное. В то же время 6-месячная терапия ингибиторами тирозинкиназы приводит к уменьшению опухоли на 21% и сокращению уровня норметанефринов и хромогранина А в плазме [90] (рис. 4).
Рисунок 4. Двусторонняя феохромоцитома и поликистоз почек.
Цистаденомы эпидидимуса — доброкачественные опухоли, которые могут быть двусторонними [48]. Чаще они имеют около 2 см в диаметре, могут распространяться на семенной канатик, приводя к бесплодию. Хирургическое лечение обычно не проводят; необходимо наблюдение [92].
У 35—75% пациентов с синдромом VHL имеются доброкачественные кисты и микрокистозные аденомы поджелудочной железы [93—96]. По данным КТ, у 17% пациентов выявляются панкреатические нейроэндокринные опухоли [95, 97, 98]. Из 633 пациентов с VHL-синдромом у 108 (17%) обнаруживались панкреатические эндокринные опухоли, и у 9 (8%) были выявлены метастазы [99]. Метастазирование более вероятно, если размеры опухоли превышают 3 см. У 78% пациентов с метастазами (7 из 9) мутация локализовалась в экзоне 3 гена VHL, и удвоение массы опухоли в среднем происходило за 337 дней. Таким пациентам необходимо оперативное лечение. Если размеры опухоли меньше 3 см, она растет медленно, а мутация локализована не в экзоне 3, то можно ограничиться наблюдением за пациентом. Средний возраст диагностики нейроэндокринных опухолей поджелудочной железы — 35 лет. Панкреатические кисты встречаются у больных начиная с 15 лет и чаще являются бессимптомными. В зависимости от размера и местоположения клинические симптомы могут быть вызваны обструкцией желчных путей и/или ферментной недостаточностью. В таких случаях устанавливают желчный стент и/или назначают ферментные препараты.
Гормонально-активные нейроэндокринные опухоли встречаются редко [95, 100]. По данным последних исследований, смертность при панкреатических эндокринных опухолях составляет 6%. В 60% случаев при сцинтиграфии обнаруживают рецепторы соматостатина; злокачественные опухоли выявлялись в 58% случаев [101].
Опухоли внутреннего уха располагаются в лабиринте, под твердой мозговой оболочкой на задней поверхности пирамиды височной кости [102—104]. Эти опухоли практически не метастазируют [105]. Симптомы включают потерю слуха, звон в ушах, головокружение и/или парез лицевого нерва [106, 107]. Таким образом, пациентам с мутацией VHL абсолютно показаны аудиологический осмотр, а также КТ или МРТ внутреннего уха с высокой разрешающей способностью, так как раннее хирургическое вмешательство способно сохранить слух. У пациентов с двусторонними опухолями, приводящими к глухоте, слух может быть восстановлен кохлеарным имплантом [108].
У 90% носителей мутации VHL к 60-летнему возрасту имеются те или иные клинические проявления синдрома [45]. На долгосрочный прогноз и смертность обычно влияет наличие гемангиобластом сетчатки и ЦНС, а также карциномы почки на поздних стадиях [31—33]. Таким образом, своевременное обследование и выявление патологии, ассоциированной с VHL-синдромом, является залогом успешного лечения и увеличения продолжительности жизни пациента (табл. 4).
Гемангиобластома ЦНС
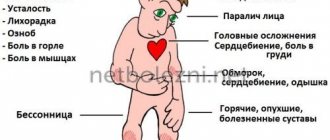
Наиболее часто регистрируемыми проявлениями синдрома фон Гиппеля-Линдау является развитие гемангиобластомы мозжечка, спинного мозга или сетчатки. Развившаяся на мозжечке опухоль имеет следующие проявления:
- Головные боли распирающего характера.
- Тошнота, рвота.
- Неуверенность при ходьбе, вследствие нарушений координации.
- Головокружение.
- Нарушения сознания (характерно для более поздних стадий патологии).
При локализации гемангиобластомы в спинном мозге, основными клиническими симптомами являются снижение чувствительности, парезы и параличи, нарушения процессов мочеиспускания и дефекации. Болевой синдром отмечается лишь изредка.
Данную разновидность опухоли относят к медленно прогрессирующим. А наиболее информативным для диагностики и контроля методом обследования признается магнитно-резонансная томография (МРТ), усиленная контрастированием исследуемого участка.
Единственным действенным способом лечения гемангиобластомы мозжечка на сегодняшний день является ее хирургическое удаление. Применение лучевого и медикаментозного методов убедительного положительного результата не показали.
После микрохирургического удаления гемангиобластома, как правило, не рецидивирует, однако могут появляться другие новообразования. При хирургическом лечении данной опухоли необходимо учитывать ее множественность, что характерно для болезни Гиппеля-Линдау.
Профилактика заболевания
Болезнь Гиппеля-Линдау травмирует людей не только физически, но и в моральном плане. Диагноз, обычно, шокирует пациентов. Такое заболевание ставит человека и его близких в чрезвычайную ситуацию. Страх перед началом болезни, страх в ожидании роста опухоли, страх перед лечением – всё это удерживает пациента почти в постоянной напряжённости.
Не нужно отчаиваться, если произнесён данный диагноз. Хотя не существует конкретных советов, чтобы избежать болезни, регулярные осмотры помогут обнаружить возникающие опухоли на ранней стадии. Пока они не вызывают никаких симптомов, как правило, хватает простого наблюдения. Дальнейшая операция может помочь избежать осложнений.
Рекомендуется ежегодно обследоваться у специалиста.
Процесс развития — стремительно и необратимо
Существует несколько стадий развития данного заболеваний:
- Доклиническая. Для нее характерны начальные скопления капилляров и небольшое их расширение.
- Классическая. На этом этапе формируются типичные ангиомы сетчатки.
- Экссудативная. Для нее характерна высокая проницаемость стенок ангиоматозных узлов.
- На четвертом этапе происходит отслойка сетчатки, причем она может иметь тракционный или экссудативный характер.
- Терминальная. На этой стадии развивается глаукома, увеит, фтизис глазного яблока.
Если начать лечение болезни Гиппеле Линдау на раннем этапе, можно добиться достаточно эффективных результатов. Кроме того, благодаря своевременной терапии удается минимизировать угрозу развития осложнений.
Для болезни Гиппеля-Линдау является характерным интенционный тремор. Какие заболевания еще сопровождает этот симптом.
Астроцитома головного мозга — серьезная опухоль, которая может привести к летальному исходу. Какие методы удаления образования существуют?
Где образуются опухоли
Как правило, туморы затрагивают центральную нервную систему, почки, глаза, надпочечники, поджелудочную железу. Реже встречаются опухоли внутреннего уха, придатков, широких связках матки.
Туморы мозжечка (гемангиобластомы) вызывают сильные головные боли, тошноту или нарушения походки. Образования в спинном мозге вызывают онемение в ограниченных участках кожи и слабость в отдельных группах мышц. Опухали сетчатки (ангиомы) могут способствовать отслоению сетчатки, повлиять на зрения и даже привести к слепоте.
Опухоли в почках и поджелудочной железе – это карциномы, поэтому являются, большей частью, злокачественными. Симптомы дают о себе знать слишком поздно, и сопровождаются уже метастазами. Туморы надпочечников могут посредством большого выброса гормонов стресса привести к головным болям, учащённому сердцебиению, потливости.
В таблице ниже представлены органы, которые могут быть затронуты этой болезнью, в процентном соотношении указано частое и редкое проявление болезни Гиппеля-Линдау.
| Орган | Проявление болезни | Процент |
| 1. Глаза | Ангиома сетчатки глаза (гемангиобластома) | 48% |
| 2. Центральная нервная система | Гемангиобластома Астроцитома | 60-80% 0,3% |
| 3. Почки | Рак почки Почечная киста | 25-45% 33% |
| 4. Поджелудочная железа | Многочисленные кисты Серозная цистаденома Опухоль островковых клеток поджелудочной железы | 17-56% 1% 8-17% |
| 5. Надпочечник | Феохромоцитома | 20% |
| 6. Ухо | Опухоль эндолимфатического мешка | 5-10% |
| 7. Придатки и широкие связки | Цистаденома и киста (у мужчин) | 50% |
| 8. Гипофиз | Аденома | 0,3 % |
| 9. Печень | Киста | 1% |
| 10. Селезёнка | Киста | 0,3% |
Стандартные методы лечения
Универсальной рекомендации по лечению не существует. Варианты лечения могут быть определены только путем тщательной оценки общей ситуации отдельного пациента — симптомов, результатов тестов, визуализационных исследований и общего физического состояния. В качестве общих рекомендаций по возможному способу лечения предлагаются следующие.
— Гемангиобластомы головного и спинного мозга.
Симптомы, связанные с гемангиобластомами головного и спинного мозга, зависят от расположения, размера опухоли и наличия связанного с ней отека или кисты. Симптоматические поражения растут быстрее, чем бессимптомные. Кисты часто вызывают больше симптомов, чем сама опухоль. Как только поражение будет удалено, киста разрушится. Если какая-либо часть опухоли останется на месте, киста снова заполнится. Небольшие гемангиобластомы, которые не имеют симптомов и не связаны с кистой, иногда лечат стереотаксической радиохирургией, но это больше профилактика, чем лечение, и отдаленные результаты, кажется, показывают лишь незначительную пользу. Кроме того, в период восстановления симптомы могут не уменьшиться.
— Нейроэндокринные опухоли поджелудочной железы.
Для дифференциации серозных цистаденом от нейроэндокринных опухолей поджелудочной железы (НЭО поджелудочной железы) необходим тщательный анализ. Кисты и цистаденомы обычно не требуют лечения. НЭО поджелудочной железы следует оценивать по размеру, поведению и конкретной генетической мутации.
— Почечно-клеточная карцинома.
Опухоли почек болезни Гиппеля-Линдау часто обнаруживаются, когда они очень маленькие по размеру и находятся на очень ранних стадиях развития. Стратегия обеспечения того, чтобы у человека была достаточно функционирующая почка на протяжении всей его жизни, начинается с тщательного наблюдения и выбора операции только тогда, когда размер опухоли или высокая скорость роста предполагают, что опухоль может получить метастатический потенциал (примерно на 3 см). При этом широко используется методика орган-сохраняющей хирургии. Радиочастотная абляция (РЧА) или криохирургия (криотерапия) могут быть рассмотрены, особенно для небольших опухолей на ранних стадиях. Необходимо соблюдать осторожность, чтобы не повредить соседние структуры и ограничить образование рубцов, которые могут осложнить последующие операции.
— Гемангиобластомы сетчатки.
Небольшие периферические поражения можно успешно лечить с минимальной потерей зрения или без нее с помощью лазера. Большие поражения часто требуют криотерапии. Если гемангиобластома находится на диске зрительного нерва, существует несколько вариантов лечения, которые позволят успешно сохранить зрение.
— Феохромоцитомы.
Хирургическое удаление проводится после адекватной блокады медикаментами, предпочтительна лапароскопическая частичная адреналэктомия. Жизненно важные показатели тщательно контролируются в течение как минимум недели после операции, пока организм приспосабливается к своей «новой норме». Особая осторожность требуется во время хирургических вмешательств любого типа, а также во время беременности и родов. Даже феохромоцитомы, которые не выглядят активными или не вызывают симптомов, следует рассматривать для удаления, в идеале до беременности или неэкстренного хирургического вмешательства.
— Опухоли эндолимфатического мешка.
Пациентам, у которых опухоль или кровоизлияние видно на МРТ, но которые все еще слышат, требуется хирургическое вмешательство, чтобы предотвратить ухудшение их состояния. Глухие пациенты с данными визуализации опухоли должны подвергнуться хирургическому вмешательству, если присутствуют другие неврологические симптомы, чтобы предотвратить ухудшение баланса. Не все опухоли эндолимфатического мешка видны при визуализации; некоторые обнаруживаются только во время операции.