Болезнь Бинсвангера – является одной из подвидов дисциркуляторной энцефалопатии — обширной группы неврологических заболеваний, обусловленных хронической гипоксией тканей головного мозга. Обычно развивается на фоне длительно прогрессирующей гипертонии, атеросклероза. Недуг сопровождается различными клиническими симптомами: ухудшением когнитивных функций, двигательными и мышечными расстройствами. Заболевание поддается терапии лишь на ранних стадиях, пока патологические изменения нервных тканей головного мозга обратимы. На поздних этапах недуга терапия сводится к поддержанию основных функций, круглосуточному уходу и контроля состояния пациента. Доктора центра психического здоровья «Лето» проведут всестороннее обследование больного, назначат правильное лечение. При необходимости возможна госпитализация в стационар нашей клиники, где человеку гарантировано должное внимание врачей и медицинского персонала, комфорт и уход врача.
Этиология и патогенез
В более чем 95% случаев основной причиной болезни Бинсвангера является гипертония. Хотя для неврологов до сих пор остается загадкой, почему одни люди страдают от прогрессирующих симптомов энцефалопатии, а у других до глубокой старости отсутствуют какие-либо изменения интеллекта, координации движений и т.д.
Предполагают, что риск развития подобного недуга особенно высок при:
- резких перепадах артериального давления (АД);
- частых гипертонических кризах;
- эпизоды гипертензии во время ночного отдыха (в норме давление должно быть ниже физиологического минимум на 10%);
- наследственной предрасположенности (инсультах, хронической гипертензии, когнитивных расстройствах у ближайших родственников);
- изменении реологических свойств крови (повышении уровня гематокрита, фибриногена, вязкости крови, что приводит к усиленной агрегации тромбоцитов на внутренней поверхности сосудистого эндотелия и сужению просвета церебральных сосудов);
- пожилом возрасте, возрастном церебральном амилоидозе;
- сахарном диабете, особенно сложно поддающимся медикаментозной коррекции;
- избыточном весе, злоупотреблении алкоголем, приеме наркотиков;
- васкулите (воспалительном процессе в сосудистом эндотелии) аутоиммунной или инфекционной природы;
- механическом сдавливании церебральных сосудов опухолью, компрессия на фоне искривления позвоночника, остеохондроза;
- гематологических патологияз (врожденных или приобретенных).
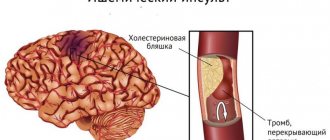
В основе патогенеза болезни Бинсвангера лежит атеросклеротическое поражение сосудистой сети, кровоснабжающей белое вещество головного мозга. Периодические колебания АД приводят к нарушениям мозгового кровообращения и лакунарным инфарктам — небольших (до 15 мм) очагов некроза.
Публикации в СМИ
В этой статье даны краткие справки о ряде болезней, не образующих отдельные статьи. См. также Синдромы разные.
Александера болезнь (203450, r) — тяжёлое нейродегенеративное заболевание ЦНС проявляющееся у детей отставанием в психомоторном развитии, судорожными припадками, параличами, мегалоэнцефалией в сочетании с обширной лейкодистрофией (распад миелина), особенно лобных долей, прогрессирующей гидроцефалией. Симптоматика подобна болезни Кэнэвэн. При обоих нарушениях отмечаются выраженные изменения астроцитов. Волокна Розенталя обычно обнаруживаются в астроцитомах, глиомах зрительного нерва и при хроническом реактивном глиозе, но при болезни Александера они особенно выражены. Лабораторно: волокна Розенталя (гомогенные эозинофильные массы, формирующие удлинённые узкие волокна до 30 мкм длиной) рассеяны повсюду в коре и белом веществе мозга, наиболее многочисленны периваскулярно и в субэпендимальных областях. Синонимы: Розенталя болезнь, Розенталя лейкодистрофия. Примечание. Гипопроконвертинемия (227500, недостаточность гемокоагуляционного фактора VII) также известна, как болезнь Александера (Alexander WS). МКБ-10. G31.8 Другие уточнённые дегенеративные болезни нервной системы.
Бало болезнь (энцефалит периаксиальный концентрический) — демиелинизирующее заболевание, клинически сходное с болезнью Шильдера (центральные параличи и парезы, гиперкинезы, эпилептиформные припадки, расстройства зрения, психические нарушения), но отличающееся наличием в белом веществе головного мозга концентрически расположенных кольцевидных участков изменённой нервной ткани. МКБ-10. G37.5 Концентрический склероз [Бало].
Бинсвангера болезнь — органическое слабоумие, развивающееся на почве атеросклероза головного мозга и церебральной гипертензии, мелкоочаговые поражения, рецидивирующий отёк белого вещества, вторичная демиелинизация « энцефалопатия Бинсвангера « Бинсвангера атеросклеротическая энцефалопатия « Бинсвангера синдром. МКБ-10. F01.8 Другая сосудистая деменция.
Венсана болезнь — острое, иногда рецидивирующее патологическое поражение дёсен, характеризующееся изъязвлением и некрозом десневого края и деструкцией десневых сосочков; обычно следствие симбиоза веретенообразной палочки (Плаута–Венсана) Fusobacterium fusiforme и спирохеты (Венсана) Treponema vincentii « язвенный гингивит « язвенно-плёнчатый гингивит. МКБ-10. A69.0 Другие инфекции, вызванные спирохетами; некротизирующий язвенный стоматит.
Гранулематозная хроническая болезнь — врождённое (формы и гены: • аутосомная, цитохром b отрицательная форма: CYBA, 233690, 16q24; • недостаточность нейтрофильного цитозольного фактора NCF 1: NCF1, 233700, 7q11.23; • недостаточность нейтрофильного цитозольного фактора NCF 2: NCF2, 233710, 1q25; • X сцепленная, CYBB, CGD, 306400, Xp21.1) нарушение переваривания фагоцитированных бактерий полиморфноядерными лейкоцитами, приводящее к снижению резистентности к инфекций (см. также Агранулоцитоз наследственный) « Врождённый дисфагоцитоз. МКБ-10. D83.0 Общий вариабельный иммунодефицит с преобладающими отклонениями в количестве и функциональной активности B-клеток.
Дарье болезнь — наследственный дерматоз (*124200, 12q23–q24.1, ген DAR, Â), характеризующийся нарушением процесса ороговения по типу дискератоза. Клиническая картина: множественные милиарные фолликулярные гиперкератотические папулы, группирующиеся в крупные бляшки с бородавчатой или папилломатозной поверхностью; умственная отсталость, возможно развитие психозов и расстройств настроения. Синонимы: Дарье вегетирующий фолликулярный псороспермоз, дискератоз фолликулярный, ихтиоз фолликулярный, кератоз фолликулярный вегетирующий. МКБ-10. L87.0 Кератоз фолликулярный и парафолликулярный, проникающий в кожу [болезнь Кирле].
Декомпрессионная болезнь — симптомокомплекс, вызванный выделением (из физиологических жидкостей) пузырьков азота (растворённого при высоком давлении); возникает при внезапном снижении давления, характеризуется головной болью, болью в руках, ногах, суставах и эпигастрии, зудом кожи, головокружением, одышкой, кашлем, удушьем, рвотой, слабостью, в тяжёлых случаях — параличами, периферическим циркуляторным коллапсом. Лечение: • 100% кислород через маску • восполнение жидкости (избегать применения 5% глюкозы и гипотонических р-ров при поражении спинного мозга) • быстрое обращение в гипербарическое отделение. МКБ-10. T70.3 Кессонная болезнь [декомпрессионная болезнь].
Демиелинизирующие болезни — группа заболеваний, морфологически характеризующихся демиелинизацией, т.е. разрушением миелиновых оболочек нервных волокон; этиология неясна (множественный склероз, острый диссеминированный энцефаломиелит, острый геморрагический лейкоэнцефалит, адренолейкодистрофия, оптиконевромиелит), врождённые нарушения метаболизма (аминоацидурии, болезнь Тэя–Сакса, синдром Гийена–Барре и др.), приобретённые нарушения (травмы, ишемия, токсические воздействия). МКБ-10. G35–G37 Демиелинизирующие болезни ЦНС • Энцефаломиелит острый диссеминированный — острое демиелинизирующее заболевание ЦНС, возникающее после вирусной, реже бактериальной инфекции (постинфекционный энцефаломиелит), вакцинации (поствакцинальный энцефаломиелит — противооспенная, антирабическая вакцины) или спонтанно и характеризующееся образованием перивенулярных очагов воспаления и демиелинизации с вариабельной общей и местной симптоматикой; лечение в основном заключается в ГК-терапии. МКБ-10. G04.0 Острый диссеминированный энцефалит • Лейкоэнцефалит острый геморрагический — острое демиелинизирующее заболевание, характеризующееся образованием перивенулярных очагов демиелинизации, воспалительной инфильтрацией, васкулитом, некрозом стенок сосудов, отложением фибрина и многочисленными кровоизлияниями в белое вещество полушарий, ствола спинного мозга и спинной мозг; клиническая картина напоминает острый диссеминированный энцефаломиелит; прогноз обычно плохой, смерть наступает в течение 2–4 дней; лечение в основном включает ГК-терапию. МКБ-10. G36.1 Острый и подострый геморрагический лейкоэнцефалит • Оптикомиелит — разновидность острого рассеянного энцефаломиелита с преимущественным поражением зрительных нервов и спинного мозга (грудные, реже шейные сегменты); клинически проявляется слепотой, центральной скотомой, очаговой симптоматикой, напоминающей множественный склероз; заболевание возникает спонтанно, после инфекционных заболеваний или как признак множественного склероза; лечение состоит в основном из ГК-терапии. Синонимы • Девика болезнь • Девика синдром. МКБ-10. G36.0 Оптиконевромиелит (болезнь Девика).
Денди–Уокера болезнь (*304340, Xq25–q27, r) — аномалии развития области IV желудочка в виде гипоплазии мозжечка (мальформация Денди–Уокера, см. статью), гидроцефалии, атрезии отверстия Люшка и Мажанди. Характерны отставание в умственном развитии, спастичность, хореоатетоз, судорожные припадки, дефекты слуха, многочисленная и разнообразная симптоматика в связи с дефектами развития головного мозга, лицевого и туловищного скелета, внутренних органов. Болезнь часто заканчивается смертью в периоде новорождённости. МКБ-10 • Q03.1 Атрезия отверстий Мажанди и Люшки • Q04.8 Другие уточнённые врожденные аномалии мозга.
Деркума болезнь. В подкожной клетчатке формируются множественные болезненные липомы и/или очаги диффузного отложение жира, встречается преимущественно у женщин (5:1) в менопаузе, сопровождается адинамией, астенией, депрессией. Синонимы • Adiposis dolorosa • Адипозалгия • Деркума синдром • Липалгия • Липоматоз болезненный • Ожирение болезненное. МКБ-10. E88.2 Липоматоз, не классифицированный в других рубриках.
Диггве–Мельхиора–Клаузена болезнь (*223800, r). Клиническая картина: диспропорциональная карликовость, выступающий подбородок, микроцефалия, выступающая грудина, бочкообразная грудная клетка, подчёркнутый лордоз, остеохондродисплазия, платиспондилия, когтеобразная кисть, общее ограничение подвижности в суставах, подвывих бёдер, нарушения походки, умственная отсталость (не всегда), сдавление спинного мозга. Лабораторные данные: гиперэкскреция мукополисахаридов с мочой. МКБ-10. Q87.1 Синдромы врождённых аномалий, проявляющихся преимущественно карликовостью.
Зайтельбергера болезнь (дистрофия нейроаксональная инфантильная, *256600, r) клинически сходна с болезнью Халлервордена–Шпатца. Отличительные признаки — кератит, задержка мочи, первичный гипотиреоз, несахарный диабет и патология терморегуляторного центра, наличие сферических телец в пределах гипоталамо-гипофизарной области и ауэрбаховского сплетения. МКБ-10. G31.8 Другие уточнённые дегенеративные болезни нервной системы.
Кальве болезнь — остеохондропатия тела позвонка. Наиболее часто поражаются нижние грудные и верхние поясничные позвонки. Клиническая картина: боли в спине, исчезающие в покое и появляющиеся при физической нагрузке, напряжение мышц спины, часто наблюдают выступание остистого отростка поражённого позвонка. Рентгенологическая картина: равномерное сплющивание тела позвонка, иногда с небольшим клином кпереди. В поздних стадиях заболевания, если лечение не проводилось, на рентгенограммах выявляют только пятнистую тень позвонка. Лечение длится несколько лет • Полная разгрузка позвоночника (постельный режим, реклинация), физиотерапия, массаж, ЛФК • Cанаторно-курортное лечение (рекомендуют пребывание в костно-туберкулёзном санатории), витаминотерапия, УФО, ношение корсета. МКБ-10. M43.0 Спондилолиз.
Кароли болезнь (*263200, мутация гена PKHD1 [600643, фиброцистин, Â]). Клинически: врождённая поликистозная дилатация внутрипечёночных жёлчных протоков, холангит, абсцессы печени, боли в эпигастрии, рвота, гепатомегалия, кровотечение из варикозно расширенных вен пищевода и желудка, портальная гипертензия, ассоциация с поликистозом почек, повторные эпизоды гипертермии. Диагностика. КТ для визуализации жёлчных ходов. Лечение • Антибактериальная терапия для снижения частоты и интенсивности эпизодов холангита • Трансплантация печени. МКБ-10. Q44.5 Врождённые аномалии жёлчного пузыря, жёлчных протоков и печени—Другие врожденные аномалии желчных протоков.
Келлера I болезнь — остеохондропатия ладьевидной кости стопы. Преобладающий возраст — 3–10 лет. Преобладающий пол — мужской. Клиническая картина • Заболевание обычно двустороннее, продолжается в течение 1 года • Боль в предплюсне, усиливающаяся при надавливании, а также ночью, иногда — припухлость • Хромота, ребёнок ходит c опорой на наружный свод стопы. Рентгенологическая картина — уменьшение костного ядра, фрагментация и уплощение ладьевидной кости, расширение суставной щели. Лечение — консервативное: ношение гипсового сапожка, физиотерапия, массаж, ЛФК. МКБ-10. M92.6 Юношеский остеохондроз предплюсны.
Келлера II болезнь — остеохондропатия головок II и III костей плюсны. Преобладающий возраст — 10–20 лет. Преобладающий пол — женский. Клиническая картина • Заболевание медленно прогрессирует, продолжается несколько лет и часто заканчивается возникновением деформирующего артроза • Боль у основания II и III пальцев стопы возникает спонтанно и усиливается во время ходьбы • В начальных стадиях часто выявляют припухлость. Рентгенологическая картина — уплотнение, пятнистый рисунок, уплощение головок плюсневых костей, расширение суставной щели. Лечение • Обеспечение покоя для конечности — иммобилизация, ношение ортопедической обуви • Хирургическое лечение — резекция головки плюсневой кости (артропластика). МКБ-10. M92.7 Юношеский остеохондроз плюсны.
Кенига болезнь — ограниченный асептический некроз суставного эпифиза. Некротизированный участок отторгается в полость сустава в виде суставной мыши. Чаще поражается коленный сустав (некроз внутреннего мыщелка бедра), реже — локтевой, тазобедренный, голеностопный суставы. Преобладающий возраст — 15–30 лет. Преобладающий пол — мужской. Клиническая картина. Выделяют две стадии заболевания: • I стадия — перемежающиеся боли, дискомфорт. Рентгенологически в области внутреннего мыщелка бедра выявляют очаг просветления с расположенным внутри костным телом • II стадия — клиника ущемления, блокады коленного сустава. Конечность остаётся в фиксированном положении, чаще в положении сгибания. В полости сустава выявляют свободную жидкость. На рентгенограммах — «пустая ниша», инородное тело в полости сустава. Лечение оперативное • В начальных стадиях рекомендуют резекцию очага остеонекроза • При отторжении фрагмента в полость сустава — удаление инородного тела • В послеоперационном периоде — иммобилизация гипсовой лонгетой, физиотерапия (УВЧ в олиготермической дозировке, токи Бернара, с 6–7 дня — ЛФК). Синоним: Рассекающий остеохондроз тазобедренного и коленного суставов. МКБ-10. M93.2 Рассекающий остеохондрит.
Кинбека болезнь — остеохондропатия полулунной кости кисти. Фактор риска — хроническая микротравматизация. Преобладающий возраст — 20–40 лет • Преобладающий пол — женский. Клиническая картина. Боли в области полулунной кости, резко усиливающиеся при надавливании и движениях в лучезапястном суставе; отёк. Рентгенологическая картина. Уплотнение, пятнистый рисунок и уменьшение полулунной кости в размерах. Лечение • Продолжительная иммобилизация, физиотерапия • Хирургическое лечение (удаление полулунной кости) показано при неэффективности консервативного лечения и продолжающихся болях. МКБ-10. M92.2 Юношеский остеохондроз кисти.
Кленового сиропа болезнь (лейциноз) — тяжёлое наследственное заболевание (ацидоз, рвота, гипогликемия, судорожные припадки, отставание умственного и физического развития, специфический запах мочи). Частота заболевания примерно 1:300 000. Биохимия: известно несколько приводящих к развитию заболевания дефектов субъединиц митохондриального ферментного комплекса — декарбоксилазы a-кетокислот с разветвлёнными цепями (КФ 1.2.4.4, формы и гены: • тип Ia: BCKDHA, MSUD1, 248600, 19q13.1 q13.2; • тип Ib: BCKDHB, E1B, 248611, 6p22 p21; • тип II: DBT, BCATE2, 248610, 1p31). При некоторых формах эффективен тиамин. МКБ-10. E71.0 Болезнь «кленового сиропа».
Кэнэвэн болезнь (*271900, КФ 3.5.1.15, мутации гена ASPA аспартоацилазы [аминоацилаза 2], 17pter p13, r) проявляется в раннем детском возрасте нарастающими параличами, слепотой; мегалоэнцефалия, выраженная демиелинизация и образование полостей в полушариях головного мозга. Клинически: атония мышц шеи, переразгибание суставов ног и рук, тяжёлое отставание в интеллектуальном развитии, мегалоэнцефалия, слепота с началом в грудном возрасте, смерть обычно в первые 18 мес. Лабораторно: губчатая дегенерация белого вещества, патология митохондрий астроцитов, увеличение в моче, ликворе и плазме крови ацетиласпартата. Синонимы: дегенерация белого вещества мозга спонгиозная, болезнь Кэнэвэн–Ван-Богарта–Бертрана, дегенерация ЦНС губчатая. МКБ-10. G37.8 Другие уточнённые демиелинизирующие болезни ЦНС.
Кюммеля болезнь — посттравматическая остеохондропатия позвонков. Клиническая картина. Выделяют три стадии заболевания • I — 5–8 дней после травмы: боль в области поражённого позвонка • II — несколько месяцев: бессимптомный период • III — упорные боли в области поражённого позвонка, кифоз. Рентгенологически: клиновидное уменьшение высоты позвонка. Лечение: разгрузка позвоночника, тёплые ванны, массаж спины, ЛФК, токи Бернара, электрофорез с хлористым кальцием, грязевые аппликации, ношение корсета. Синоним: травматический спондилит. МКБ-10. M48.3 Травматическая спондилопатия
Лафоры болезнь — миоклонус-эпилепсия (254780, локус MELF, 6q23 q25), начинающаяся в 11–18-летнем возрасте; прогрессирующие психические нарушения, на ЭЭГ изменения в задних отделах мозга, PAS положительные включения в головном мозге, миокарде, печени, коже (миоклонические тельца, или тельца Лафоры); смерть наступает в течение 10 лет от начала заболевания. МКБ-10. G40.8 Другие уточнённые формы эпилепсии.
Лева болезнь — блокада ножки пучка Хиса у больных со здоровыми миокардом и коронарными артериями; результат фиброза или кальцификации проводящей системы и вовлечения мембранозной перегородки, верхушки мышечной перегородки, колец митральных и аортальных клапанов; полиморфные и политопные экстрасистолы, обмороки; описано несколько форм. МКБ-10. I44.6 Другие и неуточнённые блокады пучка.
Ленегра болезнь — дистрофические и склеротические изменения проводящей системы сердца неясной этиологии при отсутствии поражения остальной части миокарда и венечных артерий, проявляющиеся нарушениями предсердно-желудочковой и внутрижелудочковой проводимости. МКБ-10. I45.8 Другие уточнённые нарушения проводимости.
Ли болезнь — наследственное (À, r, Â) демиелинизирующее заболевание с прогрессирующей дистрофией белого вещества головного и спинного мозга; проявляется пирамидными и экстрапирамидными нарушениями, дисфункцией черепных нервов, поражением миокарда. МКБ-10. G37.8 Другие уточнённые демиелинизирующие болезни ЦНС.
Леттерера–Сиве болезнь (*246400, r) — острый прогрессирующий гистиоцитоз у детей раннего возраста, характеризующийся лихорадкой, геморрагической пурпурой, увеличением лимфатических узлов, инфильтрацией в лёгких, гепатоспленомегалией с желтухой, панцитопенией; в красном костном мозге, печени и селезёнке значительное количество гистиоцитов « Ретикулогистиоцитоз нелипоидный. МКБ-10. C96.0 Болезнь Леттерера–Сиве.
Линдау болезнь (*193300, 3р26–p25, дефект гена VHL, Â) — множественные гемангиомы сетчатки в сочетании с гемангиомой или гемангиобластомой мозжечка и стенки IV желудочка мозга (иногда с поражением спинного мозга); возможны кисты или гамартомы почки, надпочечника и других органов. Клиническая картина • Головная боль • Прогрессирующее ухудшение зрения • Односторонняя атаксия • Головокружение • Отслойка сетчатки • Отёк диска зрительного нерва • Гемангиомы сетчатки • Субарахноидальные кровоизлияния • Полицитемия • Гемангиомы лёгких, надпочечников, мозжечка, спинного мозга, печени • Злокачественные опухоли поджелудочной железы, гемангиобластома, феохромоцитома, гипернефрома, опухоль придатков яичка • Множественные кисты почек • Гиперкальциемия и гипокалиемия при феохромоцитоме. Лечение • Хирургическое • Лучевая терапия. Синонимы • Цереброретинальный ангиоматоз • Ангиобластоматоз • Ангиофакоматоз сетчатки и мозжечка • Болезнь фон Хиппеля–Линдау • Синдром фон Хиппеля–Линдау. МКБ-10. D18.0 Гемангиома любой локализации. Примечание. Мутация гена VHL ведёт также к развитию карциномы почки.
Ляйнера болезнь (227100, r) — сочетание генерализованной эритродермии, диареи, задержки физического развития, повышенной восприимчивости к инфекциям вследствие гипоплазии лимфоидной ткани, что приводит к смерти в раннем детском возрасте.
Маркиафавы–Биньями болезнь — дегенеративный процесс в мозолистом теле, возникающий преимущественно при алкоголизме. Патогенез — прогрессирующая дегенерация мозолистого тела. Характерные признаки: прогрессирующее слабоумие, спутанность сознания, галлюцинации, тремор, ригидность и конвульсии. Течение обычно хроническое прогрессирующее. Синонимы: синдром Маркиафавы, энцефалопатия с демиелинизацией мозолистого тела. МКБ-10. G37.1 Центральная демиелинизация мозолистого тела.
Мерцбахера–Пелицеуса болезнь — наследственное (312080, PLP, PMD [липофилин], Xq22] дегенеративное заболевание головного мозга; в основе лежат прогрессирующий склероз белого вещества лобных долей и вазомоторные нарушения. Типы • Классический — нистагм и тремор появляются в первые месяцы жизни, замедленное двигательное развитие, иногда хореоатетоз, спастичность, судорожные припадки, смерть в раннем возрасте • Тип 2 — смерть в течение месяцев и десятков месяцев после рождения • Тип 3 — переходная форма, смерть в течение первого десятилетия • Тип 4 — взрослая форма. Клинически: нистагм, атрофия зрительного нерва, неонатальная гипотония, атаксия, спастические парапарезы, слабоумие, мышечные ригидность и тремор, микроцефалия, респираторный дистресс-синдром. Синонимы: диффузный семейный церебральный склероз, лейкодистрофия суданофильная, лейкодистрофия Пелицеуса–Мерцбахера. МКБ-10. G37.8 Другие уточнённые демиелинизирующие болезни ЦНС. Примечание. Мутации гена липофилина приводят также к развитию спастической параплегии типа 2 (312920).
Мачадо–Жозефа болезнь (*109150, 14q24.3–q32, ген MJD [SCA3], Â). Клинически: атаксия, паркинсонизм, дистония, мышечные фасцикуляции, отсутствие нормальных рефлексов с ног, мозжечковый тремор, разгибательные подошвенные рефлексы, экзофтальм, ограничение движений глаз, нистагм, мышечная атрофия, СД, начало после 40 лет. Лабораторно: гибель нейронов и глиоз в чёрном веществе, ядрах моста, ядрах вестибулярных и черепных нервов, колоннах Кларка и передних рогах спинного мозга, аномалии на электроокулограмме. Синонимы: спиномозжечковая атаксия типа 3, спиноцеребеллярная атрофия типа 3, азорская болезнь, спинопонтинная атрофия, нигро-спино-дентатная дегенерация. Примечание: Мачадо и Жозеф — фамилии выходцев с Азорских островов, в потомстве которых впервые описана болезнь. МКБ-10. G23.2 Стриатонигральная дегенерация.
Пайла болезнь (*265900, r). Клинически: метафизарная дисплазия, вальгусная деформация коленных суставов, бедренные кости в форме «колбы Эрленмейера», умеренное вовлечение костей черепа, аномально широкая проксимальная часть плечевой, локтевой и лучевой костей. МКБ-10. Q78.5 Метафизарная дисплазия.
Парри–Ромберга болезнь (гемиатрофия прогрессирующая лица) — постепенно развивающаяся атрофия всех тканей одной половины лица, гомолатеральная атрофия голосовой связки, гортани, языка, выпадение волос (в т.ч. ресниц и бровей). Обычно возникает в возрасте 10–20 лет. Лечение: пластическая реконструктивная операция; пересадка кожи; подкожная пересадка жировой ткани; при болях, напоминающих таковые при невралгии тройничного нерва, назначают карбамазепин. Синонимы: болезнь Ромберга, синдром Ромберга.
Периодическая болезнь — наследственное заболевание (249100, 16p13, r), наблюдаемое у армян и евреев. Клинически: кратковременные и рецидивирующие приступы лихорадки с болью в животе, груди и суставах; эритема, сходная с таковой при роже; течение рецидивирующее. Осложнение — амилоидоз, развивающийся даже при отсутствии описанных приступов. Лечение: колхицин. Синонимы: семейный пароксизмальный полисерозит, Джейнуэя–Мозенталя пароксизмальный синдром, перитонит периодический, Рейманна болезнь, Сигала–Каттана–Маму синдром. МКБ-10. E85.0 Наследственный семейный амилоидоз без невропатии.
Пика болезнь (172700). Тотальное пресенильное слабоумие с «распадом» речи — редкое прогрессирующее деструктивное заболевание головного мозга; клинически сходно с болезнью Альцхаймера, протекает с нарушениями логического мышления и восприятия, апатией, амнезией, афазией, экстрапирамидными расстройствами. Патологоанатомически: атрофия коры ограничена лобными и височными долями, необычные включения в нейронах. Течение обычно хроническое прогрессирующее. Синонимы: атрофия мозга ограниченная предстарческая, Пика синдром. МКБ-10. G31.0+ F02.0* Ограниченная атрофия головного мозга+деменция.
Пульсирующих мышц 1 болезнь (*600332; 1q41, ген RMD1, Â). Миотония с мышечной гипертрофией, отличающаяся от врождённой миотонии Томсена (160800). Клинически: миотония, мышечная гипертрофия, судороги, болезненность, ригидность, вращательные сокращения мышц (в тяжёлых случаях). МКБ-10. G71.1 Миотонические расстройства.
Рефсума болезнь — пероксисомная болезнь накопления (накопление фитановой кислоты). Генетическая классификация • Ювенильная форма (*266510, PEX1, r) • Классическая форма (*266500, PHYH, PAHX, 602026, 10pter–p11.2, r) • Болезнь Рефсума с пипеколатацидемией (*600964, r). Клинически: пигментный ретинит, миоз, птоз, атаксия, аносмия, глухота, демиелинизирующая полиневропатия, увеличено содержание белка в ликворе, изменения ЭКГ, ихтиоз; при ювенильной форме выражено отставание в умственном развитии, характерны лицевой дисморфизм (плоское лицо), гепатомегалия, стеаторея, остеопороз. Диета с исключением фитановой кислоты и плазмаферез приостанавливают прогрессирование неврологических нарушений. Синоним: болезнь накопления фитановой кислоты. МКБ-10. G60.1 Болезнь Рефсума
Семьи Байлер болезнь (*211600, 18q21–q22, дефект гена FIC1, r) — прогрессирующий семейный внутрипечёночный холестаз 1 типа. Клинически: обильный жидкий зловонный стул, эпизоды желтухи (возможно связанные с инфекцией), гепатоспленомегалия, кольцо Кайзера–Фляйшера, задержка роста, смерть между 17 мес и 8 годами жизни. МКБ-10. Q44.7 Другие врождённые аномалии печени.
Сильвершельда болезнь — хондродисплазия, характеризующаяся укорочением проксимальных и средних сегментов конечностей, утолщением эпифизов, искривлением позвоночника, карликовостью, косолапостью и седловидным носом. МКБ-10. Q77.8 Другая остеохондродисплазия с дефектами роста трубчатых костей и позвоночного столба.
Танжье болезнь (*205400, дефект посттрансляционной модификации, r) — наследуемая недостаточность ЛПВП в сочетании с низким содержанием аполипопротеина A1 (синтез последнего не нарушен), накоплением пенистых клеток, содержащих холестериновые эфиры, увеличением и яркой гиперемией миндалин, гепатоспленомегалией, лимфаденопатией, гиперхолестеринемией. Другие клинические проявления • Кардиальные — повышенный риск развития ИБС • Неврологические •• рецидивирующая невропатия •• потеря болевой и температурной чувствительности в дистальных отделах конечностей • Мышечные — прогрессирующая мышечная слабость • Офтальмологические •• снижение остроты зрения •• неполное смыкание век •• кератопатия. Синоним: Анальфалипопротеинемия. Примечание. Танжье (Tangier), остров в Чесапикском заливе (Вирджиния, США), где впервые зафиксирована болезнь. МКБ-10. E78.6 Недостаточность липопротеидов
фон Грефе болезнь — ряд наследуемых нозологических единиц в виде медленно прогрессирующей билатеральной миопатии, поражающей мышцы глазного яблока. Клинически: прогрессирующий птоз, двусторонняя наружная офтальмоплегия, слабость мышц лица, глотки, языка, плечевого пояса. Синонимы: Грефе миопатия, офтальмоплегия прогрессирующая наружная хроническая, миопатия офтальмоплегическая. МКБ-10. H49.4 Прогрессирующая наружная офтальмоплегия.
Халлервордена–Шпатца болезнь (*234200, 20p13–p12.3, дефект гена NBIA1, r) — патологический процесс демиелинизации нервных волокон, соединяющих полосатое тело и бледный шар. Клинически: афазия, двигательные расстройства, хорея, атетоз, дистония, ригидность мышц, умственная отсталость, гиперкинезы, нарушение походки, дисфагия, судороги, кривошея, тремор, деформация зубов, гримасы лица, атрофия зрительных нервов, нистагм, пигментный ретинит, блефароспазм, гиперпигментация кожных покровов, пролежни, аспирационная пневмония. Лабораторно: патология базальных ганглиев при МРТ. Лечение: препараты леводопы, триптофан, большие дозы витаминов. МКБ-10. G23.0 Болезнь Галлервордена–Шпатца.
Хенда–Шюллера–Крисчена болезнь — гистиоцитоз, характеризующийся образованием в костях, коже, лимфатических узлах, костном мозге и внутренних органах очагов пролиферации клеток (предположительно осёдлых макрофагов) с повышенным содержанием в цитоплазме эфиров холестерина; клинически проявляется несахарным диабетом, экзофтальмом, очагами деструкции костей (особенно черепа). Синонимы: Шюллера болезнь, Шюллера синдром, гистиоцитоз Х, гранулематоз липоидный, Крисчена–Шюллера болезнь, липогранулематоз. Примечание. Впервые болезнь описал в 1865 и 1876 г. английский патолог Томас Смит; Хенд А. в 1893 г. описал полиурию в сочетании, как он полагал, с туберкулёзным поражением костей; Шюллер А. в 1915 г. описал два случая поражения костей черепа при болезни Хенда–Шюллера–Крисчена; Крисчен Г. в 1919 г. привёл полное описание болезни Хенда–Шюллера–Крисчена. МКБ-10. D76.0 Гистиоцитоз из клеток Лангерханса, не классифицированный в других рубриках.
Шагаса болезнь — острая, подострая или хроническая форма трипаносомоза (возбудитель — Trypanosoma cruzi). Острые формы развиваются после укусов переносчиков — клопов-хищнецов рода Triatoma. Эпидемиология: эндемичные районы — Центральная и Южная Америка. Клиническая картина: в месте проникновения возбудителя — опухоль кожи (шагома), чаще на лице с регионарным увеличением лимфатических узлов; через 1–3 нед после укуса — инъекция склер, отёки век; лихорадка; хроническая форма характерна только для взрослых, обычно болевших в детстве (проявляется дегенеративными изменениями внутренних органов). Методы исследования: микроскопия мазков крови, серологические методы. Лечение: симптоматическое. Синонимы: южноамериканский трипаносомоз, Шагаса–Круза болезнь. МКБ-10. B57 Болезнь Шагаса.
Шейерманна–Мау болезнь — остеохондропатия апофизов (зон роста) позвонков, приводящая к искривлению позвоночника (юношеский кифоз), одно из наиболее частых заболеваний позвоночника у детей. Клинически: утомляемость, грудной кифоз, боли, усиливающиеся при выпрямлении позвоночника, надавливании. Рентгенологически: клиновидная деформация позвонков, нарушения зон роста. Лечение: постельный режим на жёсткой кровати с щитом, в острой стадии — гипсовая кроватка, ЛФК для укрепления мышц спины и живота, физиотерапия, электрофорез с хлоридом кальция, кокарбоксилазой, озокеритовые и грязевые аппликации. МКБ-10. M42.0 Юношеский остеохондроз позвоночника.
Клинические проявления
Возникающие на фоне болезни Бинсвангера лакунарные инфаркты сопровождаются:
- ухудшением тактильной чувствительности и моторики правой или левой руки;
- приступами головокружения, сильной слабости и помутнением сознания;
- головной болью.
Перечисленные симптомы носят обратимый характер. Как правило, клинические проявления постепенно нарастают в течение 3–6 дней, а затем идут на спад. Иногда лакунарные инфаркты протекают практически бессимптомно и незаметно для самого человека.
Опасность представляют повторные очаговые лакунарные инфаркты, которые и приводят к типичным для болезни Бинсвангера признакам. Это:
- Постепенное нарастание когнитивной дисфункции вплоть до развития деменции, что сопровождается потерей памяти, деградацией личности, формированием бредовых идей (как правило, преследования, ущерба, ипохондрического содержания), галлюциноза, заменой истинных воспоминаний ложными и т.д.
- Нарушения ходьбы. На начальных стадиях недуга отмечают шаркающую, неуверенную походку, раскачивание при ходьбе. По мере ухудшения состояния подобные патологические изменения прогрессируют вплоть до полной невозможности ходить и даже стоять. При этом может сохраняться чувствительность тела и конечностей, не утрачиваются и функции мозжечка.
- Обусловленное неврологической патологией периодическое недержание мочи, которое на поздних стадиях болезни трансформируется в тотальную потерю контроля над мочеиспусканием и дефекацией.
- Эмоционально-волевые расстройства: апатия, равнодушие, холодность в отношении к другим членам семьи, ухудшение интеллектуальных способностей, тяги к познанию и т.д.
- Нарушения речи и слуха из-за патологии иннервации мышц неба, голосовых связок, языка.
Начальные симптомы патологии: когда следует обратиться к врачу
- ухудшение памяти: сначала человек не может запомнить и воспроизвести недавние события и информацию, но постепенно больной начинает забывать и то, что происходило гораздо раньше;
- частые падения;
- нарушения сна;
- головная боль, сопровождающаяся звоном и шумом в ушах, нечеткостью зрения;
- слабость, снижение работоспособности, повышенная утомляемость, непереносимость физических и умственных нагрузок;
- императивные позывы к мочеиспусканию, непроизвольное отделение мочи;
Также следует обратить внимание на чрезмерную раздражительность, перепады настроения (но обычно преобладают депрессия, уныние, тоска), отвлекаемость, забывчивость. Такие проявления особенно опасны при сопутствующей гипертонии.
Формы сосудистой деменции
Мозг обеспечивает нам сознательное существование, отвечая за мыслительные, эмоциональные, адаптивные процессы. Его субстанция строго структурирована. Каждый ее отдел несет ответственность за свои функции.
Однако любой отдел мозга может подвергаться деструктивным изменениям, из-за чего нарушается определенный вид деятельности. Исходя из этого, различают несколько форм заболевания, отличающихся не только локализацией, но и калибром пораженных сосудов.
Дисмнестическая или лакунарная деменция возникает на фоне разрушения сосудов мелкого диаметра. В результате этого в толще белого и серого вещества появляются множественные инфарктные очаги. Это наиболее классический вариант течения болезни, при котором все патологические проявления неярко выражены. Наблюдается дозированное снижение интеллектуальных способностей, неярко выраженное нарушение памяти, небольшая замедленность психомоторики.
Мультиинфарктная форма сопровождается поражением сосудов среднего диаметра, и обычно развивается при неострых патологических процессах. Проявления ее незначительны и долго не замечаются даже самим больным. Течение ее поэтапное. Нарушения сначала прогрессируют, а затем застывают на определенном этапе до следующего микроинсульта. Среди симптомов этого вида заболевания на первый план выходит нарушение познавательных функций. Постепенно присоединяются неврологические и эмоциональные расстройства.
Подкорковая сосудистая деменция – это болезнь мелких сосудов, на фоне чего происходит атрофия клеток белого вещества с образованием ишемических участков. Причину этого процесса ученые видят в накоплении в стенках артерий амилоида с последующим ее воспалением. Клиника заболевания отличается некоторой нетипичностью. Она может протекать по типу болезни Альцгеймера либо в виде изолированного слабоумия.
Аутоиммунные васкулиты, такие как системная красная волчанка, панартериит становятся причиной еще одной формы заболевания – мозгового васкулита. Она выражается слабоумием и спутанностью сознания. Поражает, как правило, больных после 50.
Смешанная деменция соединяет в себе две формы: сосудистую и атрофическую, то есть по альцгеймеровскому типу. Поэтому в картине заболевания можно наблюдать симптомы как сосудистого слабоумия, так и болезни Альцгеймера, но последние превалируют над первыми.
Диагностика
Единственный достоверный способ подтвердить предполагаемый диагноз — сделать нейровизуализацию методом КТ или МРТ. На результатах томограммы отчетливо видны такие изменения:
- лейкоареоз: снижение плотности белого вещества мозга;
- множественные очаги лакунарных инфарктов;
- расширение мозговых желудочков и субарахноидального пространства.
В качестве вспомогательных методов обследования рекомендуют:
- допплерографию и/или дуплексное сканирование мозговых сосудов;
- электрокардиографию и Эхо-КГ;
- суточный мониторинг АД;
- общие клинические анализы крови и мочи (обязательна липидограмма, коагулограмма).
Дополнительно проводят психологическое тестирование для определения когнитивных и интеллектуальных функций, памяти, способности оценивать и анализировать происходящее. Также рекомендуют провести исследование на выявление маркеров наследственных заболеваний, которые могут вызвать аналогичные симптомы.
Лечение
Пациенты нуждаются в наблюдении кардиологов, неврологов, психиатров, эндокринологов, терапевтов, всех специалистов.
При болезни Бинсвангера перед лечением ставятся две цели – улучить качество жизни больного и остановить прогрессирование энцефалопатии. Больные госпитализируются, однако основное лечение проводится на амбулаторном уровне.
В основе медикаментозного лечения – восстановление нормальных показателей артериального давления и восстановление нейрокогнитивных функций. Для этого применяются Галантамин, Мемантин и Донепезил. Эмоциональная сфера восстанавливается при помощи антидепрессантов.
- Лечение назначается, исходя из стадии, степени и этиологии заболевания. Проводимая терапия направленна на этиологические и симптоматические факторы. При подборе медикаментов учитывают эмоциональное состояние, стадию основного заболевания, самочувствие пациента.
- Важный фактор в коррекции состояния пациента — контроль артериальной гипертензии. Подбор противогипертензивной терапии очень важен, так как нельзя допускать резкого снижения давления и его скачков. Давление не должно снижаться более 120/95 мм. рт. ст. так как это может привести к резкому ухудшению состояния пациента.
- Больным при атеросклеротической энцефалопатии назначают антикоагулянты и препараты, улучшающие трофику мозга. При тяжелых депрессивных состояниях пациента лечат психиатры, назначая ноотропные препараты, антидепрессанты.
- В лечении таких больных используют статины, препараты, снижающие уровень холестерина в крови. На поздних стадиях заболевания применяют палиотропную терапию. Основной задачей лечащего врача в этот период является облегчение состояния пациента.
Стоимость услуг
| КОНСУЛЬТАЦИИ СПЕЦИАЛИСТОВ | |
| Первичная консультация врача психиатра (60 мин.) | 6 000 руб. |
| Повторная консультация | 5 000 руб. |
| Консультация психиатра-нарколога (60 мин.) | 5 000 руб. |
| Консультация психолога | 3 500 руб. |
| Консультация Громовой Е.В. (50 минут) | 12 000 руб. |
| ПСИХОТЕРАПИЯ | |
| Психотерапия (сеанс) | 7 000 руб. |
| Психотерапия (5 сеансов) | 30 000 руб. |
| Психотерапия (10 сеансов) | 60 000 руб. |
| Групповая психотерапия (3-7 человек) | 3 500 руб. |
| Сеанс психотерапии у Громовой Е.В. (50 минут) | 12 000 руб. |
| ЛЕЧЕНИЕ В СТАЦИОНАРЕ | |
| Палата 4-х местное размещение | 10 000 руб./сутки |
| Палата 3-х местное размещение | 13 000 руб./сутки |
| Палата 1 местное размещение VIP | 23 000 руб./сутки |
| Индивидуальный пост | 5 000 руб. |
| ПИТ | 15 000 руб./сутки |
В данном перечне представлены не все цены на услуги, которые предоставляет наша клиника. С полным прайсом можно ознакомиться на странице «Цены», либо по телефону: 8(969)060-93-93. Первичная консультация БЕСПЛАТНА!
Описание патологии
Болезнь Бинсвангра названа по имени ученого, жившего во второй половине XIX века и изучавшего патологии головного мозга. Затем его открытия продолжил изучать Альцгеймер. Длительное время эта патология не признавалась в медицине, но все кардинально изменилось с появлением МРТ и КТ головного мозга. Благодаря современным исследованиям стало возможным проследить малейшие отклонения в строении органа.
Прогрессирующая сосудистая лейкоэнцефалопатия (синоним основного диагноза) развивается на фоне длительного повышения артериального давления в сосудах головного мозга. У таких пациентов четко прослеживается изменение количества белого вещества, которое приводит к развитию слабоумия и потере координации движений.
Болезнь тяжело поддается лечению, особенно в тяжелых случаях. Большинство пациентов с таким диагнозом получают группу инвалидности. Сегодня продолжаются изучения причин развития болезни Бинсвангера.
Для заболевания характерны нарушения сна (трудности при засыпании, беспокойный, часто прерывающийся сон), повышенная раздражительность, забывчивость, сильная утомляемость при выполнении привычной работы. Подобные нарушения являются поводом для детального обследования. Инструментальные методы позволяют выявлять характерные признаки и устанавливать диагноз с точностью 99%.
КТ-исследование показывает наличие двухстороннего лейкоареоза (поражение мелких кровеносных сосудов, пролегающих в белом веществе). При помощи МРТ обнаруживаются многочисленные диффузные участки высокой интенсивности (режим Т2) диаметром около 2 см в белом веществе. МРТ-исследование показывает характерные для болезни Бинсвангера структурные изменения в тканях:
- Уменьшение плотности белого мозгового вещества, преимущественно в области боковых желудочков.
- Наличие множественных постинфарктных кист малого диаметра.
- Отек мозга фокального или диффузного характера.
- Очаги микрогеморрагии.
- Увеличение объема и размера желудочков.
Для выявления характера нарушений в кровеносной системе проводится инструментальная диагностика методами МР-ангиографии, ультразвуковой допплерографии. Электроэнцефалография показывает особенности биоэлектрической активности отделов мозга (уровень судорожной готовности, предрасположенность к эпилептическим приступам). В ходе осмотра у невролога определяется неврологический статус пациента. Диагностические критерии:
- Деменция – основной, обязательный признак заболевания.
- Патологии сосудов в анамнезе, признаки сосудистого заболевания системного (аутоиммунного) типа.
- Признаки сосудистой патологии мозга с очаговой неврологической симптоматикой.
- Неврологические расстройства, характерные для поражения субкортикальных (подкорковых) мозговых структур – двигательные нарушения, изменение походки, паратония (неконтролируемое сопротивление пассивным движениям), недержание мочи.
При этом отсутствуют тяжелая деменция и диффузно расположенные очаги частичного некроза с локализацией в корковых структурах (по данным КТ, МРТ). Назначают анализ крови (общий, биохимический), определяют уровень глюкозы, липидов, креатинина, мочевины. При помощи электрокардиограммы выявляют особенности работы сердца (сердечный ритм, количество сокращений). Степень когнитивных нарушений выявляется по шкале MMSE. Показана консультация офтальмолога, кардиолога, терапевта.
Лечение болезни Бинсвангера в медицинском
В первую очередь подбирают антитромботическую и антигипертензивную терапию. Показатели систолического артериального давления необходимо удерживать в пределах 130–150 мм рт.ст. Наиболее безопасным препаратом для нормализации реологических свойств крови считается ацетилсалициловая кислота. Привычный многим аспирин может вызвать осложнения со стороны пищеварительного тракта, поэтому рекомендуют покрытые кишечнорастворимой оболочкой таблетированные формы в дозе 1 мг на 1 кг веса.
- Назначают препараты для:улучшения антиоксидантной функции;
- коррекции липидного профиля;
- поддержания, а в идеале — восстановления когнитивных функций;
- стимуляции мозгового кровообращения и защиты нервных клеток от гипоксии;
- коррекции мочевыделительной функции, улучшения координации походки.
Важно!
Отсутствие результата от медикаментозной терапии является показанием к хирургическому вмешательству для шунтирования сосудов.
Превентивная немедикаментозная терапия
Зная возможные факторы риска, определенные правила профилактики следует соблюдать всем пациентам с гипертензией. В первую очередь необходимо:
- Придерживаться строгой диеты, что особенно актуально при избыточном весе. Так как самостоятельно довольно сложно правильно составить рацион, рассчитать калорийность блюд, мы рекомендуем консультацию профессионального диетолога. Специалист полностью распишет меню с учетом не только состояния здоровья пациента, но и его пожеланий, ответит на все вопросы и постоянно будет на связи.
- Регулярный контроль АД. Самые точные показатели дает механический тонометр, но подобный прибор не подходит для самостоятельного использования. И если больному некому помочь, мы советуем обзавестись электронным прибором. Результаты измерений необходимо фиксировать в специальном дневнике.
- Отказ (или по крайней мере строгое ограничение) от кофеинсодержащих напитков, крепкого чая, алкоголя.
- Бросить курить, если самостоятельно преодолеть тягу к никотину не получается, мы пригласим на консультацию нарколога.
- Придерживаться режима дня, больше отдыхать, заниматься посильными физическими нагрузками.
- Массаж шейного отдела позвоночника для устранения окклюзии и улучшения кровообращения.
Как правило, для коррекции основных симптомов недуга достаточно амбулаторного медикаментозного и физиолечения. Но при необходимости (например, при прогрессировании когнитивной дисфункции, отсутствии возможности полноценно ухаживать за пациентом) наши доктора предлагают госпитализацию в оборудованный всем необходимым стационар. Для записи на прием или вызова врача на дом для консультации звоните нам по телефону 8(969)060-93-93 в любое время. При данной патологии даже день промедления может быть фатальным!
Продолжительность и качество жизни больных
Болезнь прогрессирующая, можно привести больного на более или менее нормальное качество жизни. Нужно оценить адекватность пациента, выяснить, насколько он может быть самостоятельным и трудоспособным.
По понятным причинам больных с этим заболеванием, лучше не оставлять в одиночестве или полностью обезопасить территорию, где он будет находится долго.
Желательно предусмотреть все опасные ситуации, которые возникают на пути больного человека. Чаще этих пациентов отстраняют от вождения автомобиля.
Если у человека развивается склонность к блужданию, он регулярно уходит из дома, то здесь надо предусмотреть все варианты безопасности.
Найди ответ
Мучает какая-то проблема? Введите в форму «Симптом» или «Название болезни» нажмите Enter и вы узнаете все лечении данной проблемы или болезни.
Родные такого больного должны понимать какая на них возлагается ответственность, знать всю сложность заболевания.
Заболевание ухудшает качество жизни, значительно ее укорачивает.
В плане прогноза оно неблагоприятно, но бывают яркие вспышки улучшения состояния, которые по истечении небольшого времени все равно возвращаются в исходное положение.
Хотя при правильной терапии можно продлить жизнь, поддерживая разум в сознание.
Причины возникновения
Основная предпосылка развития патологии – склеротические изменения в элементах кровеносной системы, снабжающей кровью белое вещество. Причины возникновения:
- Длительно текущая артериальная гипертензия (75-90% случаев).
- Амилоидная ангиопатия.
- Наследственная предрасположенность, в частности, аутосомно-доминантная ангиопатия, сопровождающаяся лейкоэнцефалопатией и инфарктами субкортикальной (подкорковой) локализации.
Повышенным значением артериального давления считается 140/90 мм рт. столба и больше. Факторы риска, которые способствуют развитию патологии:
- Артериальная гипотензия (у пациентов пожилого возраста).
- Скачки показателей артериального давления, связанные с нарушением циркадного ритма (существенное понижение или повышение значений в ночное время).
- Сахарный диабет в анамнезе.
- Патологии сердечно-сосудистой системы (ревматические поражения, ишемическая болезнь, патологическое изменение сердечного ритма) в анамнезе.
- Лишний вес.
Причиной развития деменции является разобщение корково-подкорковых нейронных связей, которое возникает вследствие поражения субкортикального белого вещества, нарушения функций таламуса и базальных ганглиев.
Начальные признаки
- На начальном этапе своего появления лейкоэнцефалопатия головного мозга имеет скрытое течение. Человек может представляться окружающим людям более рассеянным, неловким либо погруженным в себя. В ряде случаев отмечается повышенная слезливость, раздражительность людей. У них появляются проблемы со сном – как с засыпанием, так и прерывистость ночного отдыха.
- Дополнительно повышается мышечный тонус, что проявляется судорожными подергиваниями отдельных подгрупп мышц, движениями глаз. Возникают речевые затруднения – страдает произношение, четкость звуков. Даже короткие предложения и слава требуют приложения определенных усилий.
- Постепенно ухудшается интеллектуальная деятельность – выполнять привычные трудовые обязанности больной еще может, но при необходимости усвоить новый материал, у него появляются затруднения. Слабоумие на начальном этапе проявляющееся забывчивостью, по мере прогрессирования лейкоэнцефалопатии будет все более заметным – человек утрачивает работоспособность.
Симптомы
Клиническая картина болезни Бинсвангера проявляется психическими нарушениями. Чаще всего это расстройства интеллекта и эмоций. Постепенно снижается интеллект, вплоть до приобретенного слабоумия – деменции. Параллельно нарастают психофизиологические изменения: снижается память, рассеивается внимание.
Больной становится вспыльчивым, раздражительным. Ему трудно контролировать эмоции. В течение дня часто меняется настроение. В поведении наблюдаются странности: необоснованные поступки, социально непринятые действия и эмоциональная холодность.
По мере прогрессирования атеросклеротической энцефалопатии в клиническую картину включаются неврологические расстройства. Походка становится шаткой, больному трудно удерживать позу. Расстройство походки доходит до того, что пациент начинает внезапно падать.
Расстраивается тазовая иннервация. Больные не могут контролировать мочеиспускание, возникает эректильная дисфункция. На исходе заболевания пациенты утрачивают контроль за органами таза полностью: мочатся и совершают дефекацию «под себя».
Клиническая картина дополняется псевдобульбарными нарушениями. Возникает нарушение глотания, голос становится гнусавым, речь утрачивает четкость.