Как распознать СМА
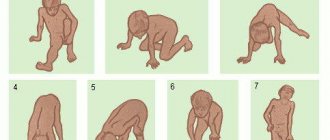
Атрофия мышц у детей при диагнозе СМА (spinal muscular atrophy) может начинаться в разное время. Самая тяжелая форма мышечной атрофии у новорожденных появляется в первые шесть месяцев жизни. Малыш вялый, плохо сосет. В 3-4 месяца ребенок не переворачивается самостоятельно, не делает попыток ползти. При спинальной мышечной атрофии поза младенца напоминает «лягушку».
Существует тип болезни, который появляется после 7-18 месяцев. Атрофия мышц приводит к регрессу у ребенка усвоенных навыков. Малыш, который ползал и начинал вставать, вдруг становится малоподвижным. Со временем он перестает сидеть ровно. При атрофии спинного мозга (spinal atrophy) пропадают рефлексы с верхних и нижних конечностей.
Симптомы атрофии мышц при spinal muscular atrophy могут возникать ближе к двум годам. Пациенты уже освоили навыки стояния и ходьбы. При этом атрофия мышц приковывает их к инвалидной коляске. Интеллект, функции мочеиспускания и дефекации сохраняются. СМА после 2-х лет – это наиболее легкий тип заболевания.
Атрофия мышц (spinal muscular atrophy) имеет следующие признаки:
- Появляется в младенчестве или детском возрасте;
- Сопровождается нарушением ходьбы, бега, стояния;
- Выявляется тремор и фасцикуляции (подергивания);
- Определяется «откат» двигательных умений;
- При спинальной атрофии не выявляют нарушений интеллекта и вегетативных функций.
Если у ребенка обнаружены эти признаки, его необходимо проконсультировать со специалистами. Диагноз СМА выставляется на основании ДНК тестирования.
IV тип
Первые признаки и симптомы появляются в 30-50 лет. Основными жалобами является слабость мышц, тремор. Из-за атрофии появляются контрактуры в области суставов, с ограничением их подвижности. Больные резко худеют.
Часто сопровождается искривлением позвоночника и деформацией грудной клетки.
Первыми в процесс вовлекаются мышцы ног, после постепенно вовлекаются руки. Как правило, дыхательная и глотательная функция не нарушена.
Большинство пациентов самостоятельно ходят, но хромают, испытывают боль и дискомфорт. В редких случаях прогрессирование амиотрофии приводит к потере способности ходить, и больные вынуждены передвигаться на инвалидной коляске.
СПИНАЛЬНАЯ АМИОТРОФИЯ ВЕРДНИГА-ГОФМАНА
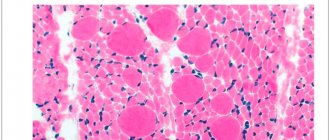
Наследуется по аутосомно-рецессивному типу. Обнаруживается недоразвитие клеток передних рогов спинного мозга, демиелинизация передних корешков, аналогичные изменения в двигательных ядрах и корешках Y, YI, YII, IX, X, XI, XII черепных нервов. В скелетных мышцах нейрогенные изменения характеризуются «пучковой атрофией», чередованием атрофированных и сохраненных пучков мышечных волокон.
КЛИНИКА
Различают три формы заболевания:
- врожденную;
- раннюю детскую;
- позднюю детскую
.
При врожденной форме
дети рождаются с вялыми парезами. С первых дней жизни выражены генерализованная мышечная гипотония и снижение либо отсутствие сухожильных рефлексов. Рано определяются бульбарные расстройства, проявляющиеся вялым сосанием, слабым криком, фибрилляциями языка, снижением глоточного рефлекса. Заболевание сочетается с костно-суставными деформациями: сколиозом, воронкообразной грудной клеткой, контрактурами суставов. Развитие статических и локомоторных функций резко замедлено. Снижен интеллект. Часто наблюдаются врожденные пороки развития: врожденная гидроцефалия, крипторхизм, гемангиома, дисплазия тазобедренных суставов, косолапость и др.
Течение быстро прогрессирующее, злокачественное. Летальный исход наступает до 9-летнего возраста. Одной из основных причин смерти являются тяжелые соматические расстройства (сердечная и дыхательная недостаточность), обусловленные слабостью мускулатуры грудной клетки и снижением участия ее в физиологии дыхания.
При ранней детской форме
первые признаки болезни возникают на втором полугодии жизни. Заболевание развивается подостро, нередко после инфекции, пищевой интоксикации. Вялые парезы первоначально локализуются в ногах, быстро распространяются на мышцы туловища и руки. Диффузные мышечные атрофии сочетаются с фасцикуляциями, фибрилляциями языка, мелким тремором пальцев, сухожильными контрактурами. Мышечный тонус, сухожильные рефлексы снижаются. В поздних стадиях возникают генерализованная мышечная гипотония, явления бульбарного паралича.
Течение злокачественное, летальный исход наступает к 14 — 15 годам жизни.
При поздней форме
признаки болезни возникают в 1,5 — 2,5 года. Заболевание начинается незаметно. Движения становятся неловкими, неуверенными. Дети часто спотыкаются, падают. Изменяется походка — они ходят, сгибая ноги в коленях (походка «заводной куклы»). Вялые парезы первоначально локализуются в проксимальных группах мышц ног, в дальнейшем сравнительно медленно переходят на проксимальные группы мышц рук, мышцы туловища; атрофии мышц обычно малозаметны вследствие хорошо развитого подкожного жирового слоя. Типичны фасцикуляции, фибрилляции языка, мелкий тремор пальцев, бульбарные симптомы — фибрилляции и атрофия языка, снижение глоточного и небного рефлексов. Сухожильные рефлексы угасают на ранних стадиях болезни. Костно-суставные деформации развиваются параллельно основному заболеванию. Наиболее выражена деформация грудной клетки.
Течение злокачественное, но мягче. Больные живут до 20 — 30 лет.
Диагностика.
Аутосомно-рецессивный тип наследования, раннее начало, наличие диффузных атрофий с преимущественной локализацией в проксимальных группах мышц, генерализованная мышечная гипотония, фасцикуляции, фибрилляции языка, отсутствие псевдогипертрофий, прогредиентное, злокачественное течение, данные электромиографии и морфологии скелетных мышц, выявляющие денервационный характер изменений.
Спинальная мышечная атрофия (амиотрофия) Верднига-Гоффмана — наследственное злокачественное заболевание, начало развития которого приходится от момента рождения до 1-1,5 года. Это одна из самых тяжелых форм мышечных атрофий. Происходит диффузное нарастание атрофии мышц по всему организму. Ребенок теряет возможность сидеть, самостоятельно передвигаться, прогрессируют парезы.
Впервые болезнь описали ученые Вердниг и Гоффман. Они доказали морфологическую сущность спинальной амиотрофии. Но они предполагали существование лишь одной формы заболевания. Позже другие ученые Веландер и Кукельберг описали другую форму спинальной атрофии мышц. Все варианты заболевания имеют одну генетическую природу. Сегодня не существует методов, которые позволяют полностью излечиться от этой патологии. Терапевтические мероприятия направлены на улучшение трофики мышц и нервной ткани.
Лечение
Основная цель исследований, направленных на терапию спинальной мышечной амиотрофии, связана с повышением уровня белка SMN. В настоящее время лекарственные препараты проходят испытания, и официальная российская медицина их не использует.
Лечение сегодня включает лекарства, которые улучшают прохождение нервных импульсов. Назначаются ноотропные препараты, основная задача которых – улучшение работы головного мозга. Назначаются биологически активные добавки, способствующие улучшению обмена веществ. Показана витаминотерапия, в частности, прием витаминов группы Б.
Средства влияющие на нервно-мышечную проводимость:
- Альфа-липоевая кислота
- Ацетил Л-карнитин
- Альфа-глицерофосфохолин
Витамины и витаминные комплексы:
- Тиамин (B-1)
- Пиридоксин (B-6)
- B-комплекс
Важными методами лечения являются массаж, физиотерапия, нейромышечная стимуляция. Назначается ЛФК. Физические упражнения помогают поддержать силу, с другой стороны, выполнение их в обществе, походы в бассейн помогают социализироваться, общаться с другими людьми.
Больным СМА рекомендовано соблюдение диеты. Продукты питания – источник веществ, необходимых мышцам. Так, необходимые аминокислоты содержатся в зерновых, мясе, рыбе, грибах, орехах, кисломолочных продуктах. Рекомендованы блюда из овса и пшеницы, бурого риса.
Советуем изучить — Симптомы эффективное лечение остеопороза позвоночника, диагностика заболевания
Естественному поддержанию и росту мышц поможет шпинат, брокколи, сельдь, лук, грейпфрут, арбуз. Для повышения тестостерона мужчинам рекомендуют принимать укроп, пастернак, женьшень, петрушку.
Лечение
Практический справочник пациента по правовым основам оказания медицинской помощи больным СМАВ этом справочнике представлены статьи и материалы по правовым основам оказания медицинской помощи и лекарственного обеспечения пациентов, требования к оформлению медицинской документации и ее формы, ответы на наиболее часто задаваемые вопросы. Фонд «Семьи СМА»
Интеллект при болезни СМА остается полностью сохранным, он развивается так же, как у здоровых людей. Таким образом, несмотря на физические ограничения, дети могут жить полноценной жизнью: общаться, играть, заниматься физиотерапией, гулять.
На данный момент заболевание является неизлечимым. Специалисты разных стран работают над препаратами для лечения СМА, но пока что общедоступных лекарств, способных полностью излечить болезнь, не существует.
В конце 2021 года в мире появился первый препарат для лечения СМА – Спинраза (Нузинерсен). Это лекарственное средство позволяет замедлить прогрессирование болезни, а в некоторых случаях и улучшить состояние, но все же не излечивает заболевание полностью. В данный момент Спинраза еще не зарегистрирована на территории России – это дело ближайшего будущего.
Кроме того, сейчас в разработке сразу несколько инновационных препаратов для лечения СМА, которые находятся на стадии клинических испытаний. Подробную и актуальную информацию об этом можно найти на сайте Фонда «Семьи СМА» в разделе «Исследования».
Тем не менее уже сейчас мы можем сделать многое, и с помощью симптоматической терапии и разнообразных поддерживающих методик замедлить прогрессирование болезни и в некоторых случаях предупредить развитие осложнений.
Стандартные методы лечения
Лекарства, которое могло бы вылечить болезнь Верднига-Гоффмана нет. Лечение направлено на определенные симптомы, которые присутствуют у каждого больного. Для лечения может потребоваться команда специалистов.
Нусинерсен является антисмысловым олигонуклеотидом, направленным на выживание моторных нейронов-2 (SMN2), и одобрен FDA для лечения СМА у взрослых и детей. Его вводят интратекально. Он увеличивает включение экзона 7 в рибонуклеиновую кислоту-мессенджер SMN2 (мРНК) и способствует производству полноразмерного белка SMN.
— Поддерживающее лечение.
Лекарства, которые часто используются для улучшения симптомов, включают фенилбутират, вальпроевую кислоту, альбутерол и гидроксимочевину. К сожалению, клинические испытания не показали каких-либо определенных доказательств того, что эти лекарства предотвращают прогрессирование заболевания. Лечение направлено на контроль симптомов. Симптоматическое лечение направлено на поддержку при кормлении, дыхании и двигательной слабости.
Проблемы с кормлением.
Дети часто испытывают трудности с кормлением, у них может быть недостаточность питания или аспирационная пневмония, вызванная затруднением глотания. Трубки для чрескожной эндоскопической гастростомии (ПЭГ) могут помочь с питанием.
Проблемы с дыханием.
Детям может потребоваться неинвазивная поддержка аппарата искусственной вентиляции легких на начальном этапе, поскольку болезнь поражает дыхательные мышцы. По мере ухудшения симптомов им может потребоваться трахеостомия и искусственная вентиляция легких.
Двигательная слабость.
Физиотерапия и трудотерапия могут помочь в растяжении, укреплении мышц и минимизации контрактур. Могут быть полезны хирургические процедуры и подтяжки, помогающие при сколиозе.
Диета при спинальной амиотрофии
На данный момент еще не подтверждено, что какая-либо диета приносит пользу при СМА.
По мнению большого количества родителей, диета, включающая в себя много белка или специальные добавки к пище, могут повысить силу мышц ребенка. Но, несмотря на очевидную необходимость хорошего питания для больного ребенка, еще не доказано, что ему нужен именно определенный рацион. Причем некоторые продукты даже могут нанести вред его организму.
К примеру, аминокислотное меню порой чревато еще большими проблемами у тех детей, у которых в организме слишком мало мышечной ткани. По мнению некоторых специалистов, при недостатке мышечной ткани, та не может правильно перерабатывать аминокислоты и тогда их уровень в крови повышается слишком сильно.
Врачами не доказано, что какая-либо диета улучшит состояние больного СМА, однако правильное питание может облегчить его жизнь.
Некоторым детям полезнее питаться понемногу, причем чаще трех-четырех раз в день. Нужно просто разделить для больного все количество пищи, принимаемое здоровым сверстником больного за день, на несколько частей.
Принципы лечения спинальной амиотрофии
К сожалению, это неизлечимое наследственное заболевание. На современном этапе проводятся исследования, которые, возможно, помогут регулировать синтез белка SMN, но результатов пока нет.
Облегчить состояние больным со спинальной амиотрофией помогают:
- периодический курсовый прием препаратов, улучшающих метаболизм нервной ткани и мышц (Церебролизин, Цитофлавин, Глутаминовая кислота, АТФ, Карнитина хлорид, Метионин, Калия оротат, Токоферола ацетат и др.);
- витамины группы В (Мильгамма, Нейровитан, Комбилипен);
- анаболические стероиды (Ретаболил, Неробол);
- средства, улучшающие нервно-мышечную проводимость (Прозерин, Нейромидин, Галантамин, Дибазол);
- курсы массажа и лечебной физкультуры;
- физиотерапия (электростимуляция мышц, углекисло-сульфидные ванны);
- методы ортопедической коррекции (при развитии контрактур суставов и деформации позвоночника).
Спинальная амиотрофия Верднига–Гоффмана, как и другие формы этого заболевания, является патологией, передающейся по наследству. Появление болезни у ребенка объясняется наличием мутантного гена и у матери, и у отца. Болезнь характеризуется, в основном, мышечной слабостью, которая становится причиной обездвиженности и дыхательных нарушений. Заболевание, на сегодняшний день, неизлечимо.
Лечебные методики
Лечение спинальной амиотрофии симптоматическое и направлено на стабилизацию состояния пациента.
Назначают лекарственные средства:
- улучшающие метаболизм – церебролизин, липоцеребин, аминалон;
- влияющие на трофику мышечной ткани – оротат калия, глютаминовая кислота, метионин, токоферола ацетат;
- способствующие нервно-мышечной проводимости – прозерин, галантамин, дибазол;
- стимулирующие кровообращение в капиллярах – компламин, никотиновая кислота;
- поддерживающие жизнеспособность двигательных нейронов – вальпроевую кислоту, рилузол, L-карнитин.
Больным предписывают ортопедические процедуры в сочетании с теплыми ваннами, показаны лечебная гимнастика, мягкий массаж, оксигенотерапия, сульфидные ванны.
Диагностика
Проявления проксимальной спинальной амиотрофии часто напоминают течение других неврологических и врожденных заболеваний, а также травматических повреждений структур спинного и головного мозга. Особенно затруднена диагностика этого заболевания у новорожденных и детей раннего возраста.
Ключевыми моментами в диагностике спинальной амиотрофии являются следующие исследования:
- Тщательный сбор анамнеза. Наличие случаев спинальной амиотрофии у родственников позволяет заподозрить это наследственное заболевание.
- Электронейромиография – специальное исследование нервно-мышечного аппарата. При этом исключается первичное поражение мышц и выявляются признаки, указывающие на патологию двигательных нейронов спинного мозга.
- Компьютерная и магнитно-резонансная томография. Эти методы позволяют иногда выявить атрофические изменения передних рогов спинного мозга. Однако, чаще они применяются для исключения другой патологии со стороны структур позвоночного столба и головного мозга.
- Биопсия мышц и с последующим гистологическим исследованием биоптата. Выявляются специфические изменения мышц, заключающиеся в чередовании пучковых атрофических и неизмененных мышечных волокон. Помимо этого, могут выявляться и компенсаторно гипертрофированные участки мышц, а также замещение мышечной ткани соединительной.
- Генетический анализ. Позволяет выявить точную причину заболевания: при исследовании ДНК выявляется мутация гена в пятой хромосоме.
Если в семье были случаи рождения детей со спинальной амиотрофией, при планировании последующей беременности супружеская пара направляется на консультацию к генетику. Также обязательной является дородовый анализ ДНК плода. Выявление синдрома Верднига-Гоффмана на стадии пренатальной диагностики служит показанием к прерыванию беременности.
Диагностика и лечение болезни Верднига-Хофмана
На ранних стадиях заболевания бывает трудно дифференциировать заболевание, так как симптоматика может быть схожа с другими болезнями:
- острый полиомиелит отличается отсутствием прогрессирования заболевания и несимметричными параличами;
- миопатия – также имеет наследственное происхождение, имеет прогрессивное течение, но причиной слабости мышц является нарушение в них обменных процессов;
- врожденная миатония наиболее схожа с болезнью Верднига-Хоффмана, отличить их достаточно можно с помощью биопсии мышечной ткани.
Для диагностики заболевания неврологу понадобятся данные о первом проявлении симптомов, характере их развития, наличия сопутствующих заболеваний.
Проводится ряд исследований для постановки диагноза:
- Электронейромиография выявляет нарушения в работе нервно-мышечной системы. Наблюдаются изменения по мышечному типу, что указывает на патологию двигательного нейтрона;
- Генетический анализ выявляет мутацию гена SMN;
- Биохимия крови на уровень креатинкиназы, показатели в пределах нормы не исключают заболевание;
- Биопсия мышц для морфологического исследования, которое выявляет пучковую атрофию мышечных волокон, чередующихся со здоровыми, а также разрастание соединительной ткани;
- МРТ для исключения других заболеваний.
Для проведения диагностики плода внутриутробно применяется метод биопсии хориона, кордоцентез, амниоцентез. Выявление заболевания является показанием к прерыванию беременности. Вылечить пациента с болезнью Верднига-Хоффмана невозможно. Для продления жизни и улучшения ее качества применяют симптоматическое лечение. Развитие болезни и ухудшение симптомов сдерживают путем обеспечения работы обменных процессов в мышечной ткани.
С помощью лечебной физкультуры и массажа улучшается кровообращение, снижается риск застоев, поддерживается работоспособность мышц, предотвращается неподвижность суставов и потеря ими эластичности. Нагрузки должны быть непродолжительными и осторожными. Физиотерапия способствует удержанию двигательных навыков на имеющемся уровне, их укрепление. Специальные приспособления помогут самостоятельно передвигаться, пользоваться компьютером и даже писать. Портативные аппараты ИВЛ дают возможность пациентам находиться за пределами стационара и проживать жизнь продуктивнее.
Спинальная амиотрофия Верднига-Гоффмана – лечится ли эта болезнь?
Спинальной амиотрофией Верднига-Гоффмана называется наследственная патология нервов, которая поражает тот их участок, что контролирует скелетные мышцы. Ее характеризует слабость сразу всех мышц в организме. Более половины нервных клеток, которые контролируют мышцы, располагаются в спинном мозге.
Именно потому заболевание названо «спинальным». «Мышечным» оно также названо из-за пагубного влияния на мышцы: те не получают от этих самых нервов никаких сигналов. «Атрофия» является медицинским термином, который обозначает истощение или усыхание того, что не используется. Здесь это касается недействующих мышц.
Человек с подобной болезнью не имеет никакой возможности ни сидеть, ни передвигаться, ни даже обслуживать себя. Вылечить это невозможно. Проведя дородовую диагностику, можно предотвратить рождение малыша с таким заболеванием.
Здесь мы поговорим, как эта патология наследуется, каковы ее проявления, а также о том, как можно помочь больному человеку.
Советуем изучить — Лордоз шейного отдела позвоночника: лечение, симптомы, диагностика и причины появления патологического нарушения
Спинальная амиотрофия Верднига-Гоффмана названа в честь двоих ученых, первыми ее описавших. Во второй половине XIX столетия они доказали морфологическую сущность болезни. Сначала оба ученых считали, что у данной патологии только одна форма.
Но уже в XX столетии ученые Кукельберг и Веландер открыли другую ее клиническую форму, с генетической причиной, схожей с той, которая и у открытой Верднигом и Гоффманом. Сейчас известно несколько клинических форм спинальной амиотрофии.
Их объединяет общий наследственный дефект.
Эта патология наследственная. Она основана на мутации в пятой хромосоме. Мутирует тот ген, благодаря которому синтезируется белок SMN. Данный белок необходим для того, чтобы двигательные нейроны развивались именно так, как нужно.
Стоит пятой хромосоме мутировать, и это негативно скажется на двигательных нейронах, помешав их развитию, а то и вовсе их разрушив. В результате мышца не может получать управляющие сигналы от нервов, а значит, не может и функционировать.
Получается, не выполняется ни одно связанное с ней движение.
У мутировавшего гена аутосомно-рецессивный тип наследования. Фраза расшифровывается так: для развития спинальной амиотрофии нужно, чтобы у обоих родителей был мутантный ген.
Если проще, то заболевание не разовьется, если хоть один из родителей не был носителем мутировавшего гена. В то же время они сами не болеют: у людей гены парные, а у отца и у матери ребенка доминирует здоровый ген.
В таком случае больной малыш рождается примерно в четверти случаев. Ученые подсчитали, что около 2% живующих людей — носители гена с такой мутацией.
Классификация
Известны три разновидности данной патологии.
- Самая тяжелая, проявляющаяся раньше остальных.
- Среднетяжелая.
- Самая легкая, проявляющаяся в самом позднем возрасте.
По мнению некоторых врачей, существует еще одна разновидность: умеренная/мягкая СМА, которая проявляется уже у взрослого человека.
Стоит отметить, что кроме спинальной амиотрофии Верднига-Гоффмана существуют и другие типы СМА, которые отличаются симптомами и типами наследования. Они указаны в таблице ниже.
| SMAX1 | Х -сцепленный рецессивный | Наблюдается в основном y пожилых, поражает бульбарные нервы чepeпa, вызывает нисходящий паралич. |
| SMAХ2 | Х — сцепл. рецессивный | Врождённая агрессивная форма, приводящая к смерти до 3 — х мес. Вызывает слабость, арефлексию, контрактуры и переломы. |
| SMAX3 | Х — сцепл. рецессивный | Поражает в основном мальчиков. Атрофия всех дистальных мышц. Медленное нарастание симптомов. |
| Дистальная ДCMA1 | Аутосомно — рецессивный | Врождённая, поражаются в основном руки, возможны тяжелее дыхательные нарушения. |
| Дистальные формы ДCMA2 — ДCMA5 | Аутосомно — рецессивный | Все четыре формы отличаются медленным прогрессированием, ДCMA5 диагностируется y молодых. |
| Ювенильная SMA (тип HMN1) | Аутосомно-доминантный | Встречается в юности |
| Врождённая спинальная aмиoтpoфия | Аутосомно-доминантный |
Причины
Спинальная мышечная атрофия связана с генетической мутацией в гене SMN1.
Человеческая хромосома 5 содержит два почти идентичных гена в местоположении 5q13: теломерную копию SMN1 и центромерную копию SMN2. У здоровых людей ген SMN1 кодирует белок двигательных нейронов, который, как следует из его названия, играет решающую роль в выживании этих клеток.
У людей, пораженных СМА, ген SMN1 мутирует таким образом, что он не может правильно кодировать белок SMN – это связано с «вырезанием» некоторых кодирующих фрагментов, или из-за других точечных мутаций (часто приводящих к функциональному преобразованию SMN1 последовательность в SMN2). Однако почти все люди имеют по крайней мере одну функциональную копию гена SMN2 (большинство из которых имеют 2–4 из них), которая все еще кодирует небольшие количества белка SMN — около 10–20% от нормального уровня. Именно поэтому некоторые нейроны все равно выживают.
Однако в долгосрочной перспективе снижение доступности белка SMN приводит к постепенной гибели клеток двигательных нейронов в переднем роге спинного мозга и в головном мозге. Мышцы, которые зависят от этих моторных нейронов, имеют уменьшенную иннервацию (также называемую денервацией) и, следовательно, гораздо меньший контроль со стороны ЦНС.
Снижение передачи импульса через двигательные нейроны приводит к снижению сократительной активности денервированной мышцы. Следовательно, денервированные мышцы претерпевают прогрессирующую атрофию из-за отсутствия их вовлеченности в движение.
Мышцы нижних конечностей обычно поражаются первыми, за ними следуют мышцы верхних конечностей, позвоночника и шеи, а в более тяжелых случаях – легочные и жевательные мышцы.
Серьезность симптомов болезни в целом связана с тем, насколько хорошо остальные гены SMN2 могут компенсировать потерю функции SMN1. Это частично связано с количеством копий гена SMN2, присутствующих в хромосоме. В то время как здоровые люди несут две копии гена SMN2, у людей с СМА может от 1 до 4 копий гена, причем чем больше количество копий SMN2, тем менее выраженное заболевание. Таким образом, большинство детей со СМА типа I имеют одну или две копии SMN2; люди со СМА II и III обычно имеют как минимум три копии SMN2; и люди со СМА IV обычно имеют по крайней мере четыре копии. Однако корреляция между серьезностью симптомов и количеством копий SMN2 не является абсолютной, и, похоже, существуют другие факторы, влияющие на выраженность заболевания.
Спинальная мышечная атрофия наследуется по аутосомно-рецессивному типу. Это означает, что дефектный ген находится на аутосоме (неполовая хромосома). Две копии дефектного гена (по одной от каждого родителя) необходимы для наследования расстройства: родители могут быть носителями и не иметь признаков болезни. Вследствие мутаций без очевидного наследования от родителей болезнь появляется только в 2-4% случаев.
Больные братья и сестры обычно имеют очень похожую форму болезни. Тем не менее, встречаются разные типы СМА среди братьев и сестер, что большая редкость.
Что такое СМА?
СМА (спинальная мышечная атрофия) ― генетическое нервно-мышечное заболевание, поражающее двигательные нейроны спинного мозга и приводящее к нарастающей мышечной слабости. Заболевание носит прогрессирующий характер, слабость начинается с мышц ног и всего тела и с развитием заболевания доходит до мышц, отвечающих за глотание и дыхание. При этом интеллект больных СМА абсолютно сохранен.
В зависимости от тяжести симптомов выделяют 3 основных типа проксимальной СМА: СМА 1, СМА 2, СМА 3. Чем раньше проявляются первые признаки болезни, тем ярче выражены симптомы, тем они тяжелее и тем быстрее прогрессирует заболевание.
СМА I (БОЛЕЗНЬ ВЕРДНИГА-ГОФФМАНА)
Наиболее тяжелая форма. Возраст проявления болезни: до 6 месяцев.
Описание
- Выраженная мышечная гипотония; синдром «вялого ребенка»; не держит голову; не достигает способности сидеть и переворачиваться; обвисшее тело при удерживании подвешенным на животе;
- Ослабленные кашлевой, сосательный и глотательные рефлексы; поперхивание; дыхательные нарушения;
- В анамнезе может быть сниженная внутриутробная активность плода. Может наблюдаться деформация суставов и конечностей из-за внутриутробной гипотонии.
Течение
- Грубая задержка моторного развития;
- Быстрое развитие контрактур и деформаций грудной клетки;
- Прогрессирование бульбарных и дыхательных нарушений, проблемы с глотанием еды и слюны, отхождением мокроты;
- Высокий риск развития аспирационных пневмоний;
- Быстрое нарастание дыхательной недостаточности, особенно при присоединении инфекции.
Прогноз
- Наиболее тяжелая форма: при отсутствии респираторной поддержки большинство детей не доживают до 2 лет;
- Смерть наступает, как правило, из-за нарастания дыхательной недостаточности и развития пневмоний;
- Своевременная респираторная поддержка может увеличить продолжительность жизни ребенка;
- Такие дети нуждаются в паллиативном наблюдении.
СМА II (БОЛЕЗНЬ ДУБОВИЦА)
Возраст проявления болезни: 6-18 месяцев.
Описание
- Отставание в моторном развитии;
- Способность сидеть без поддержки, иногда ― ползать или стоять, но эти возможности редуцируются по мере взросления;
- Может наблюдаться тремор пальцев;
- Мышечные и скелетные деформации;
- Нарушения дыхания.
Течение
- Задержка моторного развития, его остановка и регресс;
- Слабость межреберных мышц, поверхностное диафрагмальное дыхание, ослабление кашлевой функции, со временем развитие дыхательной недостаточности;
- Повышенный риск осложнений после респираторной инфекции;
- Деформации грудной клетки, контрактуры, сколиоз.
Прогноз
Своевременная помощь и респираторная поддержка увеличивают продолжительность жизни.
Юлия Самойлова. Самый известный в России человек со СМА. Фото: https://www.instagram.com/jsvok/
СМА III (БОЛЕЗНЬ КУГЕЛЬБЕРГА-ВЕЛАНДЕРА)
Возраст проявления болезни: после 18 месяцев
Описание
- Способность ходить самостоятельно (со временем теряется);
- Сложности с комплексными моторными навыками (например, подъем по лестнице, бег);
- По мере прогрессирования болезни могут отмечаться трудности с жеванием и глотанием, а также дыхательные проблемы и проблемы с откашливанием.
Течение
- Прогрессирует медленно;
- К подростковому возрасту большинство больных садятся в коляску, но у некоторых способность самостоятельно ходить может сохраниться до взрослого возраста;
- Со временем появляются выраженные контрактуры и сколиоз;
- Риск осложнений после респираторной инфекции.
Прогноз
При надлежащем уходе имеют обычную продолжительность жизни.
Симптомы
На сегодняшний день известно 4 формы спинальной амиотрофии. Все они отличаются по сроку возникновения заболевания, некоторым симптомам и длительности жизни. Общим для всех форм является отсутствие чувствительных и умственных нарушений. Функции тазовых органов никогда не страдают. Все симптомы связаны только с поражением двигательной сферы.
Спинальная амиотрофия I типа
Дебют болезни в возрасте до 6 месяцев имеет крайне неблагоприятный прогноз.
Возможны нарушения сосания и глотания, затрудненные движения языка. На самом языке могут быть видны фасцикуляции (непроизвольные мышечные сокращения, «волны» бегают по языку), а сам он выглядит атрофированным. Крик ребенка вялый и слабый. Если снижается глоточный рефлекс, то возникают проблемы с кормлением, в результате чего пища попадает в дыхательные пути. А это вызывает аспирационную пневмонию, от которой ребенок может погибнуть.
Поражение диафрагмы и межреберных мышц проявляется нарушением акта дыхания. Вначале этот процесс компенсирован, но постепенно дыхательная недостаточность усугубляется.
Характерно то, что мимические мышцы лица и мышцы, отвечающие за движения глаз, не поражаются.
Такие дети отстают в моторном развитии: они не держат голову, не переворачиваются, не тянутся за предметом, не сидят. Если какие-то двигательные навыки и смогли реализоваться до начала заболевания, то они будут утрачены.
Кроме двигательных нарушений заболевание характеризуется деформацией грудной клетки.
Если признаки заболевания видны сразу после рождения, то такие дети часто погибают в течение первых 6 месяцев жизни. Если признаки появляются после 3 месяцев, то срок жизни несколько больше – около 2-3 лет. Неизбежно присоединяется инфекция вследствие дыхательных нарушений, от которой такие детки и умирают.
Спинальная амиотрофия может сочетаться с врожденными пороками развития: олигофренией, маленьким черепом, пороками сердца, врожденными переломами, гемангиомами, косолапостью, неопущением яичек.
Спинальная амиотрофия II типа
Эта форма заболевания возникает в промежутке между первыми 6 месяцами и 2 годами жизни. До этого у ребенка не выявляют никаких нарушений. Он вовремя начинает держать голову, переворачиваться и сидеть, а иногда и ходить. А затем постепенно появляется мышечная слабость. Обычно все начинается с мышц бедер. Потихоньку становится невозможной ходьба, снижаются и утрачиваются сухожильные рефлексы. Мышечная слабость прогрессирует медленно. Вовлекаются все конечности. Развивается атрофия мышц. Процесс может захватывать и дыхательную мускулатуру. Также, как и при I-м типе спинальной амиотрофии, не поражаются мимические мышцы и мышцы глаза. Возможно дрожание кистей, подергивания на языке и конечностях. Слабость мышц шеи проявляется свисанием головы.
Весьма характерными являются костно-суставные деформации: сколиоз, воронкообразная грудная клетка, вывих тазобедренного сустава.
Эта форма имеет более доброкачественное течение, чем спинальная амиотрофия I типа, но у большинства больных к подростковому периоду имеются дыхательные нарушения. Плохая экскурсия грудной клетки способствует присоединению инфекций, от которых ребенок может погибнуть.
Спинальная амиотрофия III типа
Эта форма описана Кукельбергом и Веландером. Считается юношеской спинальной амиотрофией. Начало заболевания между 2 и 15 годами.
Первым симптомом всегда становится неустойчивая ходьба из-за нарастающей слабости в ногах. Тонус в ногах снижается, развивается мышечная атрофия (мышцы истончаются), но это не всегда заметно из-за хорошо развитого к этому возрасту слоя подкожной жировой клетчатки. Дети спотыкаются, падают, неловко передвигаются. Постепенно движения в ногах становятся невозможными, и больной перестает ходить.
Исподволь заболевание захватывает и верхние конечности, кисти рук поражаются позже. При этой форме развивается слабость мимических мышц, но движения глаз сохраняются в полном объеме. Отсутствуют рефлексы с тех мышечных групп, которые уже вовлечены в процесс.
Также характерны деформации скелета: воронкообразная грудная клетка, контрактуры суставов.
Эта форма заболевания при проведении поддерживающей терапии позволяет больным жить до 40 лет.
Спинальная амиотрофия IV типа
Эта форма заболевания считается «взрослой», потому как проявляется после 35 лет. Также возникает слабость в мышцах ног, снижение рефлексов, атрофии мышц, что, в конце концов, приводит к полной утрате движений в ногах. При этом дыхательная мускулатура не вовлекается в процесс, нарушений дыхания не бывает. Продолжительность жизни при этой форме заболевания почти такая же, как и у здоровых людей. Течение наиболее доброкачественное по сравнению с другими формами.
Диагностика
На раннем развитии заболевание бывает тяжело поставить точный диагноз, так как признаки похожи с другими болезнями. Первым делом ребенок должен пройти консультацию у невропатолога. Если у малыша заболевание при рождении, то примерный диагноз могут поставить в роддоме. Специалист проводит осмотр и проверяет нарушения двигательной системы.
Проводятся следующие исследования:
Магнитно-резонансная томография назначается для проверки позвоночника. Она обязательно используется при спинальной амиотрофии, потому как позволяет понять, в каком состоянии находится интересующая область. Процедура считается безопасной, потому как она не ухудшает состояние здоровья человека. Более того, она рекомендована даже для детей, у которых имеется амиотрофия Верднига. Если специалисты и родители понимают, что несовершеннолетний не сможет неподвижно лежать около часа, тогда может возникнуть вопрос о применении наркоза. В любом случае, от МРТ не стоит отказываться, она позволит узнать немало полезной информации о состоянии здоровья и о развитии амиотрофии Верднига.
Электронейромиогафия помогает изучить состояние нервных и мышечных окончаний. Это тоже требуется для того, чтобы понять, насколько тяжело проходит заболевание.
Если у человека спинальная амиотрофия, тогда важно как можно больше собрать информации о состоянии организма. В частности придётся проходить данное обследование.
Генетическая диагностика дает возможность выявить мутацию гена. Это актуально для тех случаев, когда имеется амиотрофия Верднига. Конечно, обследование не самое простое, и не во всех клиниках его могут провести. При этом оно является обязательным для тех людей, которые столкнулись со спинальной амиотрофией.
Найти врожденную патологию можно еще до рождения малыша. Проводится диагностика в том случае, если у девушки наблюдается слабое шевеление плода. Тогда беременная должна лечь в больницу для полного обследования.
Болезнь Фацио-Лонде
Это особый вариант проявления атрофии. Патология начинает развиваться, как правило, к трем годам жизни, и в некоторых случаях в подростковом возрасте. Для недуга характерна слабость мышц лица, в том числе жевательной мускулатуры. Отмечается затруднение глотания и изменения голоса. Патология сопровождается атрофией языка, в некоторых случаях может появиться офтальмоплегия. Прогрессирует болезнь очень быстро. Спустя 6-12 месяцев наступает смерть. К бульбарным нарушениям могут добавиться параличи и парезы в конечностях. В некоторых случаях эти симптомы не успевают даже развиться. Тем не менее, при вскрытии всегда выявляется поражение в клетках передних спинномозговых рогов на всем протяжении.
Причины и факторы возникновения болезни
Почему появляется спинальная амиотрофия? Какие факторы риска можно выделить? Какова главная причина недуга? Болезнь носит генетический характер и передается аутосомно-рецессивным путем. При этом дефектный ген должен быть у обоих родителей. В этом случае спинальная атрофия у ребенка возникает в 25% случаев.
Как гены влияют на развитие болезни? Спинальная амиотрофия появляется при дефиците или при полном отсутствии белка SMN. Он обеспечивает выживаемость двигательных нейронов. Его недостаток – главная причина атрофии мышц при СМА. Клетки мозга умирают, и от них не идет сигнал к мышцам.
При делеции длинного плеча 5-ой хромосомы, на которой расположен ген SMN 1, белок не вырабатывается. Развивается спинальная амиотрофия.
Факторы риска:
- Семейный анамнез отягощен по мертворождениям;
- Болезнь выявлена у близких родственников;
- Семейные случаи младенческой смертности;
- Спинальная амиотрофия у старшего ребенка.
Вероятность возникновения мышечной атрофии у младших детей 1:4.
Что нужно запомнить?
- Спинальная амиотрофия – редкая генетическая патология, приводящая к инвалидности и смерти пациента.
- Основная причина атрофии мышц – генетическая мутация.
- I тип болезни – амиотрофия Верднига-Гоффмана наиболее тяжелая и частая амиатрофия, приводящая к смерти ребенка.
- Основной метод диагностики – генетическое тестирование на обнаружение дефектного гена.
- Это заболевание неизлечимо.
- Прогноз на выздоровление и качество жизни – неблагоприятный.
Литература
- А.Н. Бакланов, С.В. Колесов, И.А. Шавырин. Хирургическое лечение тяжелых нейромышечных сколиозов у пациентов, страдающих спинальной мышечной атрофией // Хирургия позвоночника. — 2011. — № 3.
- Селиверстов Ю.А. Спинальные мышечные атрофии: понятие, дифференциальная диагностика, перспективы лечения /Ю.А. Селиверстов, С.А. Клюшников, С.Н. Иллариош-кин // Нервные болезни. — 2015. — № 3.
- Hardart M.K. Spinal muscular atrophy-type I /M.K. Har-dart, R.D. Truog//Arch. Dis. Child. — 2003. — Vol. 88, № 10.
- Wilton N.C. Spinal muscular atrophy: the challenges of «doing the right thing» / N.C. Wilton // Paediatr. Anaesth. — 2009. — Vol. 19, № 11.
- The changing natural history of spinal muscular atrophy type 1 / M. Oskoui, G. Levy, C. J. Garland // Neurology. — 2007. — Vol. 69, № 20.
- Anesthesia and perioperative medical management of children with spinal muscular atrophy / R..J. Graham, U. Athiraman, A.E. Laubach // Paediatr. Anaesth. — 2009. — Vol. 19, № 11.
Виды спинальных амиотрофий
Спинальная мышечная атрофия – одно из самых опасных генетически обусловленных заболеваний, которое обнаруживается у младенцев, подростков, взрослых.
Страшно узнать, что малыш никогда не будет сидеть, стоять, бегать. Еще страшнее видеть, как нормально растущий и развивающийся ребенок вдруг начинает медленно угасать, постоянно падать, через несколько месяцев не может подняться по лестнице, а однажды теряет способность просто встать.
Условно различают проксимальные и дистальные формы СМА. 80% всех видов спинальных амиотрофий относятся к проксимальной форме.
К ним относятся, кроме заболевания Верднига-Гофмана:
- СМА 3 или болезнь Кульдберга-Веландер — заболевают в возрасте от 2 лет до 20, первыми страдают мышцы таза. Отмечается тремор кистей, лордоз.
- Летальная X-сцепленная форма — описана в 1994 году Baumbach, наследуется по рецессивному признаку, наблюдаются преимущественно поражения мышц таза и плечевого пояса.
- Инфантильная дегенерация — нарушаются рефлексы сосания, глотания, дыхание. Смерть может последовать в возрасте до 5 месяцев.
- СПА Рюкю — ген сцепливания не выявлен, наблюдается отсутствие рефлексов, мышечная слабость конечностей после рождения.
К дистальным спинальным амиотрофиям относится прогрессирующий детский паралич Фацио-Лонде, болезнь Брауна-Виалетта-ван Лэре, СМА с параличом диафрагмы, эпилепсией и глазодвигательными нарушениями.
Прогноз: сколько живут пациенты?
Спинальная амиотрофия крайне тяжелое заболевание, которое существенно ухудшает качество жизни больных и является одной из причин младенческой смерти. Пациенты с таким диагнозом являются инвалидами и зачастую не в силах самостоятельно себя обслуживать.
Многие пациенты не могут ходить и стоять, передвигаясь с помощью инвалидной коляски. При поражении рук больные не способны делать элементарных вещей: есть, держать небольшие предметы, читать, умываться и прочее. Они нуждаются в дополнительном уходе и постоянном медицинском наблюдении. При тяжелом течении они дышать лишь с помощью аппарата ИВЛ и питаются через зонд.
Продолжительность жизни во многом зависит от формы заболевания. Наиболее тяжелой является амиотрофия Верднига, при которой больные дети редко достигают 4 лет. Пациенты со вторым типом болезни редко доживают до совершеннолетия. Наиболее благополучным являются второй и третий тип болезни, при которых пациенты достигают зрелого возраста.
В настоящее время спинальная амиотрофия относится к неизлечимым заболеваниям, часто приводит к летальному исходу.
Клиническая картина
Признаки СМА 1 и СМА 2 сильно различаются. Проявления СМА 1 нередко выявляют ещё во время беременности, так как плод в утробе матери неактивно и очень редко шевелится. После рождения у ребёнка отмечается дыхательная недостаточность, нередко он не может сам дышать, отсутствуют и другие врождённые рефлексы. Основными проявлениями болезни можно считать:
- Пониженный мышечный тонус.
- Отставание в физическом развитие.
- Неспособность удерживать голову в вертикальном положении.
- Не может самостоятельно перевернуться на бок, на живот или на спину.
- Слабо выраженный паралич конечностей.
- Отсутствие рефлексов глотания.
- Отсутствие рефлекса сосания.
Ребёнок принимает характерную позу, в которой находится практически постоянно. При этом в большинстве случаев может отмечаться и паралич диафрагмы. Всё это со временем приводит к нарушению развития скелета, появляется сколиоз, нередко отмечается наличие горба, изменяется форма грудной клетки. Только 12% таких детей доживают до 5 лет.
I тип амиотрофии
Наиболее злокачественный и распространенный тип патологии – амиотрофия Верднига-Гоффмана. Диагностируется у детей раннего возраста либо внутриутробно. У детей после рождения отмечается снижение и угасание всех рефлексов. Им сложно сосать грудь, из-за чего часто возникает необходимость кормить ребенка через зонд.
Мышечная гипотония приводит к слабости шейных мышц – дети не могут научиться держать головку, самостоятельно переворачиваться, сидеть, стоять и ходить. У большинства детей возникают трудности с глотанием. В большинстве случаев амиотрофию диагностируют в течение 6 месяцев после рождения. Родители обращают внимание на вялость ребенка и малоподвижность, прогрессирующее снижение мышечного тонуса. Также младенцы плохо набирают вес.
Характеризируется поражением диафрагмы, которая участвует в дыхательном акте. Многие пациенты самостоятельно не дышат. Из-за выраженной дыхательной недостаточности многие дети не доживают до года. Продлить им жизнь удается с помощью ИВЛ и питания через зонд. 95% детей умирают до 4 лет.
Патология сопровождается прогрессирующими деформациями опорно-двигательной системы.
Помимо поражения мышц и костей, часто выявляется отставание в умственном развитии.
Справка. Заподозрить диагноз можно еще во время беременности матери, когда отмечается слабое шевеление плода, нарушение сердечной деятельности, отставание в развитии.