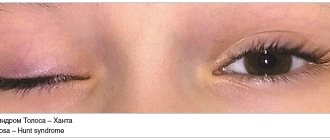
Guillain-Barré syndrome is a severe autoimmune disease that affects the peripheral nervous system. The most common manifestation is acute tetraparesis, when movement of all four limbs becomes almost impossible. Other movements also cease, including swallowing, the ability to lift the eyelids, and spontaneous breathing. Despite this, the course of the disease is benign, most cases end in recovery. Transition to a chronic course or relapses are less common. Guillain-Barré syndrome occurs in all countries, regardless of their level of development, with the same frequency - approximately 2 cases per 100 thousand population, no dependence on gender. The disease can affect patients of all ages.
At CELT you can get advice from a neurologist.
- Initial consultation – 4,000
- Repeated consultation – 2,500
Make an appointment
Why does the syndrome occur?
The leading mechanism of development is autoimmune. In most cases, the onset of the disease occurs in the first three weeks after an acute respiratory or intestinal infection. Since a sufficient amount of time has passed since the moment of illness, and the symptoms characteristic of the infectious process have time to pass, the patients themselves, as a rule, do not associate these conditions with each other. The cause may be pathogens such as:
- Epstein-Barr virus or human herpes type 4;
- mycoplasma;
- Campylobacter, which causes infectious diarrhea;
- cytomegalovirus.
Researchers have found that the “sheath” of these pathogens is similar to the myelin sheath of the axon of peripheral nerves. This similarity causes the nerves to be attacked by antibodies, which are initially produced and circulate in the blood in response to the appearance of an infectious agent. This phenomenon is called “molecular mimicry” and explains why immune complexes attack the body’s own tissues.
Cases have been described when the syndrome occurs after vaccination, after surgery and abortion, hypothermia, and stress. In some cases, the cause cannot be found.
How does the syndrome manifest?
Over the course of several days, up to a maximum of 1 month, muscle weakness in the legs increases, and difficulties arise when walking. Next, the hands weaken, the last to suffer is the facial muscles. Such symptoms have a separate name - ascending Landry's paralysis.
But sometimes the paralysis begins from above, from the arms, spreading downwards, but all limbs are always affected.
Every fifth case is accompanied by paralysis of the trunk muscles, namely the diaphragm and intercostal muscles. With such paralysis, breathing becomes impossible and artificial ventilation is required.
A common manifestation is bulbar syndrome or bilateral paralysis of the muscles of the soft palate, when swallowing and clear speech are impossible.
Together with motor fibers, sensory fibers are sometimes affected. Sensory disturbances develop, tendon reflexes decrease, and pain in the limbs occurs. The pain is of a pronounced “neuropathic” nature - burning, a feeling of current passing, tingling. Pelvic disorders are rare, but most often there is urinary retention, which in some cases is combined with excess urine production.
Autonomic dysfunction is added, which is manifested by fluctuations in blood pressure, palpitations, other heart rhythm disturbances, sweating, and lack of intestinal motility.
Diagnostic measures
Diagnosis is based on WHO recommendations. There are two leading diagnostic criteria:
- muscle weakness in the limbs that progresses;
- decreased or absent tendon reflexes from the first days of the disease.
WHO also identifies additional signs that confirm the diagnosis, which include:
- symmetry of the lesion;
- symptoms increase within no more than 4 weeks;
- sensory disturbances of the “gloves and socks” type;
- involvement of the cranial nerves, especially the facial nerve;
- possible spontaneous restoration of functions after stopping the progression of the disease (the so-called “plateau”);
- the presence of vegetative disorders;
- absence of hyperthermia (if there is fever, it is caused by other infections);
- an increase in the amount of protein in the cerebrospinal fluid, while its cellular composition does not change (protein-cell dissociation).
Definitive diagnosis is impossible without electroneuromyography or ENMG. This test reveals which part of the nerve is damaged - the myelin sheath or the axon. ENMG also accurately determines the extent of the lesion, its severity and the possibility of recovery.
Since, in addition to Guillain-Barré syndrome, there are a number of acute, subacute and chronic polyneuropathies, electroneuromyography allows for differential diagnosis between them and contributes to the development of correct treatment tactics.
Diagnosis often requires a lumbar puncture followed by a study of the cerebrospinal fluid, and tests such as:
- blood for autoantibodies to neuronal structures;
- blood for class A gammaglobulins (especially if immunoglobulin therapy is planned);
- biomarkers of neurofilament (part of the cytoplasm of a neuron);
- markers of tau protein (a special protein that destroys neurons).
Specialists at the CELT clinic additionally use their own differential diagnostic algorithms, which make it possible to reliably distinguish Guillain-Barré syndrome from other diseases that cause progressive muscle weakness in all extremities or tetraparesis.
Diagnostics
- Careful collection of anamnesis for previous infections and vaccinations.
- EMG is not always informative, as it is often within normal limits in the first weeks. Typical changes are:
- in acute inflammatory demyelinating polyneuropathy: decreased impulse conduction speed along motor nerves, proximal type conduction block, increased latency of F-waves.
— about acute sensorimotor axonal polyneuropathy: a decrease in the amplitude of the total action potentials of motor muscles and sensory nerve fibers, signs of demyelization in a maximum of 1 nerve.
- in acute motor axonal polyneuropathy: a decrease in the amplitude of the total action potentials of the motor muscles, sensory fibers remain intact, demyelination is also in a maximum of 1 nerve.
- in acute sensory polyneuropathy: motor fibers are not affected, there is a decrease in the amplitude of the total action potential of the sensory nerves.
- Study of cerebrospinal fluid: protein-cell dissociation. In the first days, however, it is not always possible to detect it, therefore the study of the cerebrospinal fluid should be carried out over time. It should be remembered that false-positive results are possible with intravenous administration of immunoglobulins.
- MRI of the brain and spine allows you to see changes in the roots, but to a greater extent this method is used in the differential diagnosis of tumors and stroke.
- Blood tests: detection of antibodies in the blood
- for acute inflammatory demyelinating neuropathy - antibodies unknown
— for acute motor axonal neuropathy – GM1+GM1b, GD1a
- for acute motor-sensory axonal neuropathy - GM1+GM1b, GD1a
- for acute sensory neuropathy - GD1b
- for Miller-Fisher syndrome - GQ1b.
Our doctors
Pankov Alexander Rostislavovich
Neurologist
40 years of experience
Make an appointment
Novikova Larisa Vaganovna
Neuropathologist, Candidate of Medical Sciences, doctor of the highest category
Experience 39 years
Make an appointment
Treatment rules
Today, two main pathogenetic methods of treating Guillain-Barré syndrome are known, and both are successfully used by CELT specialists. These are plasmapheresis and intravenous immunotherapy. These methods can be used in isolation or used in combination, it all depends on the specific clinical situation. Treatment is aimed at removing or neutralizing immune complexes circulating in the patient’s blood. Both treatment methods are equivalent and almost always lead to recovery. Treatment stops the process of destruction of peripheral nerves, shortens the recovery period, and helps reduce neurological deficits.
Plasmapheresis is a blood purification operation. Most often, hardware plasmapheresis is used on continuous separators, during which the blood taken from the body is divided into formed elements (or blood cells) and plasma (or serum). All toxic substances are in the plasma, so it is removed. The person is given back his own blood cells, diluted, if necessary, with plasma-substituting solutions or donor plasma. The duration of the procedure is about one and a half hours, the entire course consists of 3 or 5 sessions. No more than 50 ml/kg of plasma body weight is removed at a time.
During treatment, blood parameters are monitored: electrolytes, hematocrit, clotting time and others.
Intravenous immunotherapy is the administration of a human immunoglobulin class G drug. These immunoglobulins stop the production of antibodies to one’s own nerves, simultaneously reducing the production of substances that support inflammation. These drugs are indicated for the pathogenetic treatment of Guillain-Barré syndrome in both adults and children.
Simultaneously with specific treatment, careful patient care is provided, including the prevention of bedsores, pneumonia, and contractures. Treatment of concomitant infections is often required. Venous thrombosis is prevented, tube feeding is carried out, and excretory function is monitored. Bedridden patients undergo passive exercises, as well as early verticalization to avoid blood flow disorders. If there is a threat of developing contacture (immobility of joints), paraffin procedures are possible. If necessary, motor simulators based on biofeedback are used.
Patients with damage to myelin sheaths recover faster, while axonal damage requires a longer period of rehabilitation. Axonal lesions often leave behind neurological deficits that are difficult to correct.
Guillain-Barré syndrome: etiology, clinical picture, treatment
Guillain-Barré syndrome (GBS) is an acute inflammatory demyelinating polyradiculoneuropathy of autoimmune etiology, characterized by peripheral paralysis and, in most cases, protein-cell dissociation in the cerebrospinal fluid.
The first mention of the disease dates back to the 18th century. In 1859, Landry J. described acute ascending paralysis. In 1916, French doctors Guillain G., Barre J. and Strohl A. described the clinical picture of acute peripheral paralysis with protein-cell dissociation of the cerebrospinal fluid in two French soldiers. In 1949, Haymaker WE and Kemohan JW described the clinical picture and histological changes in the peripheral nervous system in 50 patients with GBS [9].
The incidence rate of GBS is 0.6-2.4 cases per 100 thousand population. GBS occurs with equal frequency among men and women [3,10,23]. According to data presented by Suponeva N.A. et al. (2013) in 7 cities and 2 constituent entities of the Russian Federation, the incidence of GBS ranged from 0.34 to 2.5 per 100 thousand population [4]. In Moscow, about 200 people become ill with GBS every year [6].
The development of the disease is preceded by contact of the body with a viral or bacterial infection. According to the literature, symptoms of this disease appear 10-14 days after a viral respiratory infection [10,33]. Infectious agents can include pathogens such as Campylobacter jejuni, Mycoplasma pneumonia, cytomegalovirus, Epstein-Barr virus and influenza virus. Cases of GBS after surgery and vaccinations have been described in clinical practice [9,33].
GBS is characterized by an autoimmune lesion of the peripheral nervous system, which most often leads to demyelination and then to secondary axonal destruction of nerve fibers, sometimes damage to myelin and axons can occur simultaneously. GBS is based on autoimmune mechanisms that are triggered as a result of a viral or bacterial infection. Infectious agents on their surface have structures (oligosaccharides) similar to the structures of peripheral nerves, resulting in “molecular mimicry” [5]. As a result of “molecular mimicry,” the production of autoantibodies to antigens of the peripheral nervous system occurs [5].
In the acute phase of the disease, a change in cellular and humoral immunity occurs. Antibodies to peripheral nerve myelin are detected in the blood serum of patients, T-cell activity increases and the number of T-suppressors decreases. As a result of activation of humoral and cellular immunity, the concentration of IgM, IgA and circulating immune complexes (CIC) increases. This leads to the accumulation of CEC along the myelin sheaths of peripheral nerves [5,9,10].
One of the evidence of the involvement of infectious agents in inflammation is the detection in patients of a high titer of antibodies to the gangliosides GM1, GD1a, GD1b and GQ1b against the background of an increased titer of antibodies to the putative pathogen [20,28,34]. Moreover, a higher level of antibodies to ganglioside GM1 is a risk factor for the development of acute motor axonal neuropathy, which is a more severe form of GBS, manifested mainly by sensory impairment and poorer recovery [33].
The significance of “molecular mimicry” is clearly evident in patients with Miller-Fisher syndrome (MFS). “Molecular mimicry” between infection (Campylobacter jejuni) and peripheral nerve structures plays a key role in damage to axon terminals [5,32]. In patients with MFS, antibodies to gangliosides GQ1b and GT1a are detected in the blood, which target the oculomotor and bulbar nerves [18,25,26,32].
The final stage of the pathogenesis of GBS is the entry of T cells and CECs into the endoneurium along with macrophages. This leads to severe tissue damage, which is accompanied by active phagocytosis carried out by cells of the monocyte-macrophage lineage [3,20].
GBS usually begins with muscle weakness and/or sensory disturbances in the lower extremities, which then spread to the upper extremities. According to the literature, progression of the disease in 50% of patients is observed by the 2nd week from the onset of the first clinical symptoms. By the 4th week, GBS is diagnosed in 90% of patients. Approximately 80-90% of patients require hospitalization [9,10,16].
Clinical symptoms in the advanced stage of GBS usually consist of motor, sensory and autonomic disorders; tendon hypo- or areflexia. Motor disorders (paresis of the limbs of varying severity, often up to paralysis) are observed in almost all patients. In severe cases, the majority also experience damage to the muscles of the trunk, including the muscles of the neck, back, and abdomen. Muscle weakness in the extremities is usually symmetrical and more pronounced in the legs, but it may be slightly predominant on one side of the body.
A neurological examination reveals motor disorders that are symmetrical in nature. With GBS, cranial nerves (VII, IX X) can be affected, resulting in impaired swallowing, phonation and oculomotor disorders.
It must be remembered that in addition to the neurological manifestations of the disease, patients may experience complications from other organs and systems. According to Chio A. et al (2003), approximately one third of patients develop respiratory failure resulting from paresis of the diaphragm and respiratory muscles [3,14,15,33]. Patients with GBS are characterized by complications from the cardiovascular system, which manifest themselves in the form of arterial hypertension, tachycardia or bradycardia [15]. Due to impaired neuromuscular conduction, one third of patients experience bladder dysfunction, manifested in the form of urinary retention. Gastrointestinal disorders are found in 15% of patients with GBS, including such a serious complication as intestinal obstruction [9].
Currently, four main clinical variants of GBS have been described. The most common (classical) variant is acute inflammatory demyelinating polyradiculoneuropathy (85-90%) [14,23]. The axonal form of GBS and acute motor axonal neuropathy account for 10-15% of all cases of GBS [27]. These GBS variants are much more common in Asian and South American countries (30-47%) compared to European and North American countries [7]. Miller-Fisher syndrome occurs in no more than 3% of cases and is characterized by ophthalmoplegia, cerebellar ataxia with mild paresis [7,21,23].
The updated diagnostic criteria for GBS are the following: progressive motor weakness involving more than one limb, areflexia or severe hyporeflexia. To confirm the diagnosis of GBS, cerebrospinal fluid analysis and electroneuromyography are used. When analyzing cerebrospinal fluid, diagnostic criteria confirming GBS include an increase in protein concentration and the absence of cytosis. According to Ropper AH (1992), characteristic changes in the cerebrospinal fluid are diagnosed in more than 90% of patients during the height of the disease [27,28]. Electroneuromyographic study reveals a slowdown in nerve conduction velocity and late F-wave responses [9].
The North American Motor Impairment Severity Scale (NAS) is used to assess the neurological status of patients with GBS. SAS allows you to assess the patient’s condition and his motor ability from stage 0 (normal) to stage 5 (need for mechanical ventilation) (Table 1).
Table 1
North American Motor Impairment Severity Scale
| Degree | Signs |
| 0 | Norm |
| I | Minimal movement disorders |
| II | Ability to walk 5 m without support (support) |
| III | Ability to walk 5 m with support |
| IV | Inability to walk 5 m with support or support (bedridden or wheelchair bound) |
| V | The need for mechanical ventilation |
Currently, the erasmus GBR outcome score (EGBR) prognostic scale and its modification mEGBR are used in everyday practice (Table 2) [1]. The EGBR prognostic scale allows, within the first 1-2 weeks, to predict the possible restoration of walking with support by the 6th month of the disease. The EGBR prognostic scale, which was proposed by Van Koningsveld R. et.al (2007), takes into account the patient’s age, diarrhea and the degree of motor impairment. The higher the score of a GBS patient on the EGBR scale, the higher the likelihood that the patient will not be able to move independently within 6 months (Table 2) [31].
table 2
Modified Erasmus Guillain-Barré Syndrome Outcome Scores (mEGOS)
| Prognostic factor | Point |
| Age, years | |
| <40 | 0 |
| 41-60 | 1 |
| ˃60 | 2 |
| Previous diarrhea | |
| Availability | 0 |
| absence | 1 |
| MRC result upon admission | |
| 51-60 | 0 |
| 41-50 | 2 |
| 31-40 | 4 |
| 0-30 | 6 |
| mEGOS | 0-9 |
Treatment of patients with GBS has two main goals: intensive therapy aimed at preventing and relieving clinical manifestations of respiratory failure, preventing thrombosis, the addition of secondary infectious complications and suppressing the autoimmune process that leads to damage to the myelin sheath of peripheral nerves, that is, specific therapy.
Currently, specific therapy includes plasmapheresis (PP) and immunoglobulin G (IgG) therapy, which is carried out in the first weeks of the disease [7,8,34].
Plasmapheresis (PP) is one of the methods of efferent therapy, in which the volume of removed plasma is replaced by solutions of crystalloids, albumin and donor plasma. PF has been used in patients with GBS since 1985. According to the recommendations of the American Society of Apheresis (2010), PF is a standard procedure for GBS [29]. The PF regimen involves removing 200-250 ml/kg of plasma over 7-14 days. The removed plasma volume is replaced with 5% albumin. Indications for PF are increasing neurological symptoms requiring mechanical ventilation, inability to walk more than 5 m with support or support, or inability to stand and walk 5 m independently [2,8,17,19,20,30]. As a result of a series of control studies, it was found that the inclusion of PF can speed up the healing process and reduce the time of artificial ventilation [7,14,21,22,25]. As an example, we can cite the results obtained by the French Joint Group, which studied the time spent by patients with GBS on mechanical ventilation. The results obtained in this study showed that the average time that patients with GBS were on mechanical ventilation in the group where PF sessions were conducted was 18 days. In the control group, this indicator was 31 days [29].
Hughes RA et al. (2003) provides data from a comparative meta-analysis of the effectiveness of PF. According to the results obtained, in the group of patients who underwent PF, restoration of full muscle strength was noted in 135 out of 199 patients. In the control group – in 112 out of 205 patients [15]. According to an analysis conducted by the American Academy of Neurology in 2003, it was noted that PF promotes faster recovery in patients with GBS with a disease duration of 4 weeks (level A recommendation) [27].
Currently, along with PF, another method of efferent therapy has become widespread - immunosorption (IS). IS allows antibodies or antigens to be bound and extracted from blood using immunosorbents. The binding reaction of certain molecules is based on the antigen-antibody reaction. The IS method allows the removal of Ig from circulating plasma without the need to use replacement solutions such as albumin solution or fresh frozen plasma, which reduces the risk of allergic or infectious complications [8,13]. Immunosorption columns contain a sorbent gel consisting of tryptophan covalently bonded to polyvinyl alcohol. The literature provides data on the use of IS in patients with GBS [19]. The results of a comparative analysis of the effectiveness of IS and PF did not demonstrate the advantages of the first method over PF [8,19,24].
Since 1988, IgG has been used in the treatment of this disease. Intravenous administration of high doses of IgG has been recognized as an effective treatment for GBS, capable of significantly reducing the duration and severity of the disease [12]. As in the case of PF, the mechanism of the therapeutic action of IgG remains not fully understood. It is believed that immunoglobulin eliminates pathogenic antibodies, blocks the Fc component of antibodies on target cells, and also inhibits complement deposition, dissolves immune complexes, weakens the functions of lymphocytes, disrupts the production or interferes with the functions of cytokines [11,28].
For adults and children with GBS, IgG is used at a dose of 2g/kg for 2-5 days. The American Academy of Neurology (2003) recommended the use of IgG in patients with disease duration not exceeding 2 weeks (level A recommendations) [11].
An international randomized control study of the comparative effectiveness of PF, IgG and a combination of PF and IgG was conducted in 383 patients with severe GBS. The average time to increase muscle strength and the ability to walk without assistance in the group where PF was used was 49 days, in the group where IgG was used - 51 days, and in the group of combined use of PF and IgG - 40 days. This study demonstrated equal effectiveness of the two treatment options for GBS [11,29].
The use of corticosteroids is currently considered ineffective and is not used in the treatment of patients with GBS [15]. A review of six studies conducted in 587 patients demonstrated no beneficial effect of corticosteroids on the course of the disease [17,22].
According to the literature, approximately 20-30% of patients develop respiratory failure, requiring artificial pulmonary ventilation (ALV) [10,27]. Indications for transferring a patient to mechanical ventilation are an increase in respiratory failure: tachypnea, use of auxiliary muscles, tachycardia, decrease in vital capacity of the lungs to 20 ml/kg [27]. Patients on mechanical ventilation are at high risk of developing complications such as pneumonia, tracheobronchitis or sepsis. The average duration of mechanical ventilation ranges from 2 to 6 weeks [9].
Paresis and paralysis that occur in patients with GBS increase the risk of developing thromboembolic complications. To prevent deep vein thrombosis and pulmonary embolism, low molecular weight heparin is prescribed in prophylactic doses.
Most patients with GBS report pain. According to the study, about 47% of patients perceived the pain as “terrible”, “unbearable”. In this study, 75% of patients required the use of narcotic analgesics to relieve pain [9]. Currently, the drugs of choice are gabapentin, carbamazepine, and pregabalin.
Approximately 40% of patients with GBS require rehabilitation. The main group consists of patients who have undergone mechanical ventilation for a long time or with a more severe course of the disease. The complex of rehabilitation measures includes massage and physical therapy [9].
During therapy, including PF or IgG, complete restoration of motor functions is observed in the majority of patients (60-80%). However, in some patients, especially with the axonal form of GBS, there is worse recovery of motor functions [3,7]. Mortality with GBS averages 5% and can reach 20% in patients on mechanical ventilation [27,28]. The most common cause of death in patients with GBS may be respiratory failure, aspiration pneumonia, sepsis, or pulmonary embolism. Mortality increases significantly with age: in children under 15 years of age it does not exceed 0.7%. while in persons over 65 years of age it reaches 8.6%. Other unfavorable prognostic factors for a full recovery include a long period of mechanical ventilation (more than 1 month) and the presence of somatic pathology. Persistent residual symptoms persist in approximately 7-15% of cases [14,21,23,25]. Predictors of an unfavorable functional outcome are age over 60 years, rapidly progressing course of the disease, low amplitude of the M-response upon stimulation at the distal point (implying severe axonal damage). The recurrence rate of GBS is 3-5% [3,27].
Bibliography
- Grishina D.A., Suponeva N.A., Piradov M.A. Clinical prognostic factors in Guillain-Barré syndrome. Bulletin of Ross Military Medical Academy 2013;4(44):21 – 26.
- Levin O.S. Polyneuropathy. M.: Medical Information Agency; 2006.560 p.
- Piradov M.A. Guillain-Barre syndrome. M.: Intermedica; 2003.240 p.
- Suponeva N.A., Piradov M.A., Gnedovskaya E.V. Guillain-Barré syndrome in the cities of the Russian Federation: epidemiology, diagnostic and therapeutic capabilities of regional clinics Health Care of the Russian Federation 2013;1:19 – 25. II
- Suponeva N.A. Clinical and diagnostic role of autoantibodies to peripheral nerve glycosides: review of the literature and our own data. Neuromuscular Diseases 2013;1:26 – 35. III
- Suponeva N.A., Mochalova E.G., Grishina D.A., Piradov M.A. Features of the course of GBS in Russia: analysis of 186 cases. Neuromuscular Diseases 2014;1:37 – 46. I
- Suponeva N.A., Piradov M.A., Grishina D.A., Molchalova E.G. The effectiveness of pathogenetic therapy for Guillain-Barré syndrome. Effective Pharmacotherapy 2014;58:12 – 23.
- Balogun RA, Kaplan A, Ward DM et al. Clinical application of therapeutic apheresis. J Clin Apheresis 2010;25:250 – 264.
- Burns TM Guillain-Barré syndrome. Semin Neurol 2008;28(2):152 – 167.
- Chio A., Cocito D., Leone M. et al. Guillain-Barré syndrome: a prospective, population-based incidence and outcome survey. Neurol 2003;60:1146 – 1150.
- Dada MA, Kaplan AA Plasmapheresis treatment in Guillain-Barré syndrome: potential benefit over IVIg in patients with axonal involvement. Ther Apher Dial 2004;8:409 – 412.
- Donofrio PD Immunotherapy of idiopathic inflammatory neuropathies. Muscle Nerve 2003;28:273 – 292.
- Haupt WF, Rosenow F., van der Ven C., Birkmann C. Immunoadsorption in Guillain-Barré syndrome and myasthenia gravis. Ther Apher 2000;4(3):195 – 197.
- Hiraga A., Mori M., Ogawara K. et al. Recovery patterns and long term prognosis for axonal Guillain-Barré syndrome. J Neurol Neurosurg Psychiatry 2005;76:719 – 722.
- Hughes RA, Wijdiks EF, Barohn R. et al. Quality Standards Subcommittee of the American Academy of Neurology. Practice parameter: immunotherapy for Guillain-Barré syndrome: report of the Quality Standards Subcommitte of the American Academy of Neurology. Neurol 2005;61:736 – 740.
- Hughes RA, Cornblath DR Guillain-Barré syndrome. Lancet 2005;366:1653 – 1666.
- Hughes RA, Swan AV, van Koningsweld R., van Doorn PA Corticosteroids for Guillain-Barré syndrome. Cochrane Database Syst Rev 2006;2:1446.
- Kanzaki M., Kaida K., Ueda M. et al. Ganglioside complex containing GQ1b as target in Miller Fisher and Guillain-Barré syndromes. J Neurol Neurosung Psychiatry 2008;79:1148 – 1152.
- Kaynar L., Altuntas F., Aydogdu I. et al. Therapeutic plasma exchange in patients with neurological diseases: retrospective multicenter study. Transfus Apher Sci 2008;38:109 – 115.
- Lee MC, Campbell R., Born C. Guillain-Barré syndrome after failed pelvic fracture fixation. J Trauma 2009;67:132 – 135.
- Lehmann HC, Hartung HP, Kieseier BC, Hughes RA Guillain-Barré syndrome after exposure to influenza virus. Lancet Infect. Dis 2010;10(9):643 – 651.
- Levin KH Variants and mimics of Guillin Barré syndrome. Neurologist 2004;10:61 – 74.
- Mc Grogan A., Madle GC, Seaman HE, de Vries CS The epidemiology of Guillain-Barré syndrome worldwide. A systematic literature review. Neuroepidemiology 2009;32(2):150 – 163.
- Okamiya S., Ogino M., Ogino Y. et al. Tryptophan-immobilized column-based immunoadsorption fs the choice method for plasmapheresis in Guillain-Barré syndrome. Ther Apher Dial 2004;8:248 – 253.
- Overell JR, Willson HJ Recent development in Miller Fisher syndrome and related discorders. Curr Opin Neurol 2005;18:562 – 566.
- Paprounas K. Anti-GQ1b ganglioside antibody in peripheral nervous system disorders. Arch Neurol 2004;16:1013 – 1016.
- Ropper AH The Guillain-Barré syndrome. N Engl J Med 1992;326:1130 – 1136.
- Susuki K., Rasband M.N., Tohyama K. et. al Anti-GM1 antibodies cause cjmplement-mediated disruption of sodium channel clusters in peripheral motor nerve fibers. J Neurosci 2007;27(15):3956 – 3967.
- Szczepiorkowski ZM, Winters JL, Bandarenko N. et al. Guidelines on the Use of Therapeutics Apheresis in Clinical Practice-Evidence-Based Approach from the Apheresis Applications Committee of the American Society for Apheresis. J Clin Apheresis 2010;25:83 – 177.
- Tripp A. Acute transverse myelitis and Guillain-Barré overlap syndrome following influenza infection. CNS Spect 2008;13:744 – 746.
- Van Koningsveld R, Steyerberg EW, Hughes RAC et al. A clinical prognostic scoring system of Guillain-Barré syndrome. Lancet Neurol 2007;6:589 – 594.
- Willison HJ The immunobiology of Guillain-Barré syndromes. J Peripher Nerv Syst 2005;10:1094 – 1112.
- Willison HJ Ganeglioside complexes as targets for antibodies in Miller Fisher syndrome. J Neurol Neurosurg Psychiatry 2006;77:1002 – 1003.
- Yuki N. Guillain-Barré syndrome and anti-ganglioside antibodies a clinician-scientist's jorney. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci 2012;88(7):299 – 326.
Prevention
The main method is the complete cure of infections that we consider banal and habitual. Guillain-Barré syndrome often develops with a slight weakening of the immune system, which is possible in every person.
The easiest way to protect yourself is to check your current immune status. This will only take a few days, and any abnormalities detected can be treated in time.
Doctors at the CELT clinic have at their disposal not only the latest diagnostic equipment, but also the latest treatment techniques that have received worldwide recognition. The main role in prevention belongs to the patient who seeks examination and treatment in a timely manner.
Make an appointment through the application or by calling +7 +7 We work every day:
- Monday—Friday: 8.00—20.00
- Saturday: 8.00–18.00
- Sunday is a day off
The nearest metro and MCC stations to the clinic:
- Highway of Enthusiasts or Perovo
- Partisan
- Enthusiast Highway
Driving directions
Differential diagnosis
- Spinal form of poliomyelitis. At the onset of the disease there may be meningeal signs; then asymmetrical flaccid paresis and paralysis develop, a decrease in tendon reflexes only on the affected side, and there are practically no sensory and autonomic disorders. Currently, given the vaccination against polio, the disease is rare and differential. Diagnosis of this disease is becoming increasingly rare.
- Spinal cord tumor. Common with Guillain-Barré syndrome: clinical picture and changes in the CSF. Differences: it often develops gradually, paresis is spastic in nature, pyramidal signs are detected.
- Polyneuropathy in acute intermittent porphyria. It is characterized by the onset of autonomic disorders: abdominal pain, constipation and diarrhea, rapid heartbeat, increased blood pressure. Also with this pathology, mental disorders are observed, such as anxiety, depression, and in more severe cases, delirium and convulsive syndrome. Polyneuropathy is of a peculiar nature: the upper extremities are the first to be involved, starting from the proximal parts; tendon reflexes remain intact longer, and sensory disturbances occur less frequently and are milder in severity.
- Brain stem stroke. The disease often manifests itself with complaints from the head: headaches, dizziness, accompanied by nausea and vomiting, speech impairment. Then alternating syndromes are added, characterized by damage to the cranial nerve on the side of the pathological process and motor or sensory disorders of the limbs and body on the opposite side.