- How common are neuroblastomas?
- Risk factors for neuroblastoma
- What are the causes of neuroblastoma?
- Is it possible to prevent the development of neuroblastoma?
- Is early diagnosis of neuroblastoma possible?
- How is neuroblastoma diagnosed?
- Treatment of neuroblastoma in children
- What happens after treatment for neuroblastoma ends?
Neuroblastoma is a type of malignant tumor and usually occurs in infants and children and very rarely in children over 10 years of age. The cells of this tumor resemble nerve cells in the early stages of their development in the fetus.
One third of neuroblastomas develop in the adrenal glands, the other third in the abdominal cavity along the nerve trunks along the spine, and the rest in the chest cavity and neck. Some neuroblastomas arise from the spinal cord. Sometimes, due to the wide distribution of the tumor at the time of diagnosis, it is difficult to determine the exact location where the tumor originated.
Not all tumors of the nervous system are malignant.
Ganglioneuroma is an example of a benign tumor.
Ganglioneuroblastoma is a mixed tumor and contains both malignant and benign areas. Immature (malignant) cells from this tumor can grow and metastasize.
Ganglioneuromas are usually removed surgically and then carefully examined under a microscope to determine whether there are areas of ganglioneuroblastoma. If ganglioneuroma is diagnosed, no additional treatment is required. In contrast, ganglioneuroblastomas are treated in the same way as neuroblastomas.
Neuroblastoma is an unusual tumor for many reasons. This tumor secretes a hormone that leads to unusual changes in the body, for example, rotational movements of the eyeballs, spastic muscle twitching, and the presence of constant loose stools.
These changes are called paraneoplastic syndromes.
Neuroblastoma can behave in unusual ways. Sometimes tumor cells spontaneously die and the tumor disappears. This phenomenon occurs most often in very young children and much less frequently in older patients. In some cases, tumor cells spontaneously mature and stop dividing. Thus, neuroblastoma turns into ganglioneuroma, a benign tumor.
How common are neuroblastomas?
Neuroblastoma is the most common tumor in infants and is the fourth most common malignant tumor in older children, after acute leukemia, central nervous system tumors, and malignant lymphomas.
Every year, 6-8 children per 1 million children under 15 years of age (average age 2 years) become ill with neuroblastoma in Russia.
Approximately 650 new cases of neuroblastoma are diagnosed each year in the United States, a number that has remained stable for many years. Neuroblastoma occurs slightly more often in boys than in girls. For every 6 cases of tumors in boys, there are 5 cases in girls.
The average age of patients at the time of diagnosis is 17 months. One third of cases are diagnosed in children under 1 year of age. Almost 90% of neuroblastomas are detected before 5 years of age. Only 2% of tumors are detected after the age of 10 years and in adult patients. In rare cases, neuroblastoma can be detected by ultrasound before the baby is born.
In 7 out of 10 cases, neuroblastoma already has metastases at the time of diagnosis.
Risk factors for neuroblastoma
A risk factor is something that increases the likelihood of developing a tumor.
Lifestyle factors are major factors in adult cancer. Examples include an unhealthy diet (low intake of fruits and vegetables), physical inactivity, smoking and drinking alcohol. Risk factors associated with children's lifestyle do not affect the occurrence of malignant tumors.
The likelihood of neuroblastoma occurring around the world is almost the same. This suggests that environmental factors, such as pollution, are not the cause of the development of this tumor.
The only known risk factor for neuroblastoma is heredity. It is believed that some people may very rarely inherit the risk of developing neuroblastoma. Only in 1-2% of cases is the familial form of neuroblastoma diagnosed, i.e. the occurrence of a tumor in a child in whose family there were cases of this disease.
The average age of patients at the time of diagnosis of familial cases of neuroblastoma is 9 months. This is less than the age of patients with sporadic (non-inherited) cases of tumor. In addition, children with familial neuroblastoma may develop two or more similar tumors in different organs, such as both adrenal glands.
It is important to distinguish neuroblastomas, which develop simultaneously in different organs, from metastatic neuroblastoma, when the tumor arises in one organ and then spreads throughout the body. If neuroblastoma occurs in several places, then a familial form of the disease can be assumed. Neuroblastoma metastases can occur in both familial and sporadic cases of the tumor.
Treatment
As with other types of cancer, the priority in the treatment of neuroblastoma is surgery. In this case, both complete and partial removal of the tumor can be performed. If less than half of the tumor was removed during surgery, chemotherapy is performed at the second stage. A second operation is then considered again to remove the remaining neuroblastoma.
If surgical treatment is not possible, chemotherapy is prescribed. If the patient belongs to a high-risk group, he is prescribed high-dose chemotherapy and additionally undergoes a stem cell transplant. If chemotherapy is ineffective, radiation therapy is used.
The prognosis for neuroblastoma is relatively favorable.
If the tumor was detected at an early stage, the 3-year survival rate is close to 100%. At stage III of neuroblastoma this figure is 80-98%, and at stage IV - 60-76%. Book a consultation around the clock +7+7+78
What are the causes of neuroblastoma?
The exact causes of neuroblastoma are not clear. However, there are known differences between neuroblastoma cells and normal neuroblasts. The differences between neuroblastomas that respond to treatment and those that do not respond to therapy and have a poor prognosis (outcome) of the disease have been clarified. This information is important when developing treatment approaches.
Many researchers believe that neuroblastoma occurs when normal embryonic neuroblasts do not mature into nerve cells or adrenal cortical cells. Instead, they continue to grow and divide.
Neuroblasts may not be fully mature by the time the baby is born. In fact, it has been shown that small clusters of neuroblasts are often detected in infants up to 3 months of age. Most of these cells eventually mature into nerve cells and do not form neuroblastoma. Sometimes the neuroblasts remaining in infants continue to grow and form a tumor, which can even metastasize to various organs. However, many such tumors eventually mature or disappear.
As the child grows, the likelihood of these cells maturing decreases, and the likelihood of developing neuroblastoma increases. Once neuroblastoma reaches a large size and symptoms appear, the cells stop maturing and continue to grow and spread unless treated.
Some cancer patients have DNA mutations (changes) that they inherited from one of their parents, which increases their risk of developing a tumor. Some believe that some familial cases of neuroblastoma result from inherited mutations in a tumor-inhibiting gene.
Most neuroblastomas are not due to inherited DNA mutations. They are caused by mutations acquired early in a child's life. These mutations are present in the tumor cells of the child's parent and are not passed on to the children. The causes of DNA changes that lead to neuroblastomas are not known.
Is early diagnosis of neuroblastoma possible?
Studies have shown that screening (examination in the absence of symptoms of the disease) in children for the purpose of early diagnosis of neuroblastoma is of no value.
Screening (testing urine to detect certain substances) at 6 months of age has actually been able to diagnose more cases of neuroblastoma.
However, these tumors were those that either disappeared later on their own or matured and, perhaps, would never have been diagnosed.
For the same reason, it is believed that such screening will not reduce mortality from neuroblastoma.
Moreover, current screening methods are not as specific as we would like. Of the two children with suspected neuroblastoma, according to screening data, only one case actually had a tumor. These false results led to undue concern among parents and operations in children in the absence of neuroblastoma.
In rare cases, neuroblastoma can be diagnosed before birth using ultrasound (ultrasound). This method is used to determine the age of the fetus, predict the time of birth of the child and identify certain birth defects. Improvements in ultrasound and other techniques may lead to more accurate diagnosis of neuroblastoma before the baby is born.
How is neuroblastoma diagnosed?
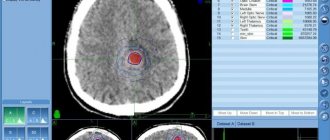
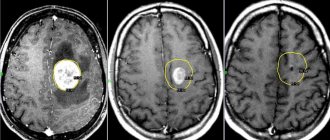
If a child has signs and symptoms suggestive of neuroblastoma, additional testing will be required, including blood and urine tests, microscopic examination of tissue, and the use of imaging techniques such as computed tomography (CT) , magnetic resonance imaging (MRI) , scanning , and etc.
This additional testing is necessary because many of the symptoms of neuroblastoma can be caused by other tumors and non-tumor diseases.
Signs and symptoms of neuroblastoma depend on the location of the primary tumor and the extent to which it has spread to nearby or distant organs and parts of the body.
The most common sign of neuroblastoma is the discovery of a tumor in the abdomen, which leads to an increase in its size. The child may complain of a feeling of abdominal distension, discomfort or pain as a result of the presence of a tumor. However, palpation of the tumor does not cause pain. The tumor may also be located in other areas, such as the neck, spreading beyond the eyeball and causing it to bulge.
Neuroblastoma often affects the bones. In this case, the child may complain of bone pain, limp, and refuse to walk. If the tumor spreads into the spinal canal, compression of the spinal cord may occur, leading to weakness, numbness and paralysis of the lower extremities.
Every fourth patient may have a fever.
Less commonly observed:
- Constant loose stools (diarrhea)
- High blood pressure leading to irritability
- Cardiopalmus
- Skin redness
- Sweating
These signs and symptoms result from the release of hormones by neuroblastoma cells.
Sometimes patients experience swelling of the lower extremities and scrotum due to compression of the blood and lymphatic vessels in the pelvic area. In some cases, a growing tumor can lead to dysfunction of the bladder and colon. Neuroblastoma's pressure on the superior vena cava, which carries blood from the head and neck to the heart, can cause swelling of the face or throat. These phenomena, in turn, can lead to breathing or swallowing problems.
As a result of pressure on nerves in the chest and neck, symptoms such as drooping eyelids and constricted pupils can occur. Compression of nerves near the spine can cause a child to lose the ability to move their arms or legs.
The appearance of bluish or reddish spots that resemble small bruises may indicate skin damage by a tumor process.
Due to the involvement of the bone marrow, which produces blood cells, a child's blood counts may all decrease, which can cause weakness, frequent infections, and increased bleeding from minor injuries (cuts or scrapes).
Normal nerve cells communicate with each other by releasing certain chemicals, the main ones being catecholamines. In the body, catecholamines are broken down into metabolites, which are then excreted in the urine.
In 90% of cases, neuroblastoma cells produce such an amount of catecholamines that can be detected in the urine using special methods. Two main metabolites of catecholamines are usually determined: homovanillic acid (HVA) and vanillylmandelic acid (VMA).
Some symptoms associated with neuroblastoma, such as high blood pressure, rapid heartbeat, and loose stools, are directly attributed to increased levels of catecholamines.
A biochemical blood test allows you to judge the function of the liver and kidneys.
Computed tomography (CT), magnetic resonance imaging (MRI), bone scans, and ultrasound (US) are used to identify the tumor and its metastases.
Although a number of examination methods suggest the presence of neuroblastoma, the final diagnosis is made only when neuroblastoma cells are detected during microscopic examination.
Sometimes neuroblastoma cells can be confused with cells from other childhood tumors. In this case, immunohistochemical examination of the tumor is used after special treatment with antibodies. These antibodies bind to neuroblastoma cells, unlike cells from other childhood tumors.
If the levels of catecholamines or their metabolites are elevated, then detection of neuroblastoma cells in the bone marrow is sufficient to establish a definitive diagnosis. The bone marrow may be affected in up to 25% of patients with neuroblastoma.
Some clinics are using a new scanning method using radioactive metaiodobenzylguanidine (MYBG). The method allows you to detect neuroblastoma cells in the bone marrow and other parts of the body.
Clinical manifestations
Like other types of malignant tumors, in the early stages neuroblastoma is asymptomatic. A neoplasm can be detected by chance when the patient is undergoing examination for another reason. Clinical manifestations appear when the tumor reaches a relatively large size and begins to compress nearby structures. At the same time, the symptoms are nonspecific and it is very difficult to suspect an oncological process. Depending on the location of the tumor, these may be:
- Constipation, bloating, vomiting, diarrhea when neuroblastoma is localized in the abdominal cavity.
- Trouble swallowing or breathing if the tumor is located in the chest area.
- Neurological disorders when the primary tumor is localized in the spinal canal.
The tumor can also be located in the pelvis, neck, or along the spinal column. In some cases, it is not possible to determine the primary location of the tumor.
Other nonspecific symptoms may include weakness, fatigue, swollen lymph nodes, poor appetite, and bone pain. These manifestations are typical for later stages of neuroblastoma, when there are already metastases to other organs and tissues. Most often, the tumor metastasizes to the bone marrow, bones, lymph nodes, liver, lungs and various parts of the central nervous system.
Treatment of neuroblastoma in children
Every child with neuroblastoma should receive treatment. The treatment method depends on the stage of the tumor, the age of the child, and prognostic factors. Treatment includes surgery, chemotherapy and/or radiation therapy. In some children, two or all three therapies may be used.
Usually, when choosing a treatment method for neuroblastoma, they take into account not so much the stage of the disease as the risk group.
Low risk group. In general, low-risk children require only surgery, with the exception of some patients. For this small group of children, chemotherapy may be used.
Intermediate risk group. Patients with this risk group are prescribed 4-8 cycles of chemotherapy before or after surgery. In some cases, repeated surgery is performed to remove the remaining tumor or radiation therapy.
High risk group. For this group of patients, very intensive chemotherapy is used along with bone marrow or peripheral stem cell transplantation.
Surgery and/or radiation therapy may be part of the overall treatment program. Biological agents, such as 13-cis-retinoid acid, are prescribed for 6 months after discontinuation of therapy.
Unfortunately, some patients after treatment for neuroblastoma may experience a recurrence (recurrence) of the disease after initial treatment. Treatment in this case depends on many factors, including risk group and location of relapse, and may include surgery and chemotherapy.
Neuroblastoma
Despite the fact that neuroblastoma has been intensively studied by clinicians and experimenters for more than 100 years, the treatment of this disease remains an important problem in pediatric oncology. The choice of treatment strategy, at least, depends on two factors: - prognostic signs identified in children during the diagnosis of the disease; - tumor response to treatment. By answering these questions, the chemotherapist, surgeon and radiologist can determine rational treatment tactics for a child with neuroblastoma, which should be intensive if the patient has negative prognostic signs, and less aggressive if the prognosis is good. Unfortunately, neuroblastoma still remains a poorly predictable tumor.
Chemotherapy Ninane (1990) provides the following antitumor drug treatment regimens for children with neuroblastoma.
In monochemotherapy of patients with neuroblastoma, carried out in usual doses, 7 drugs turned out to be significantly effective: vincristine, cyclophosphamide, cisplatin, doxorubicin, vepeside (VP-16), teniposide (VM-26) and melphalan. Later, the antitumor activity of ifosfamide and carboplatin in neuroblastoma was shown.
Before 1980, a number of chemotherapy regimens consisted of vincristine and cyclophosphamide with or without adriamycin. The results of treatment using these regimens were disappointing for children older than 1 year with locally advanced tumors, since only 10% of children survived 2 years. The addition of cisplatin, vepeside and teniposide led to an increase in the initial antitumor effect, but had little effect on long-term results.
In recent years, high-dose treatment regimens with bone marrow transplantation have been proposed to improve the results of chemotherapy).
We note the following: 1. The use of high doses of melphalan in the form of a single drug made it possible to obtain results that were not very different in effect from the results that were observed after intensive polychemotherapy or its combination with total irradiation. 2. Mortality due to the toxicity of the treatment was lowest in the group of patients who received only high doses of melphalan.
It should be noted that this treatment was carried out in the form of consolidation therapy in children with common forms of the disease. It was shown that patients with complete or significant tumor regression after initial chemotherapy had better 2-year survival rates compared with those whose tumors had little response to initial treatment.
Interesting data on long-term follow-up of patients with neuroblastoma who underwent intensive chemotherapy were presented by Philip et al. (1990) and Dini et al. (1990).
The first researchers observed, on average, 55 months of 62 patients with neuroblastoma in stage IV, whose age was more than 1 year. Children in the entire group received intensive consolidation chemotherapy with bone marrow transplantation. 40% of children lived for two years without disease progression, 25% for 5 years, and 13% for 7 years. However, in the group of patients with complete regression of metastases, 37% of children lived for 2 and 7 years. Dini et al. (1990) provided data on 34 patients with stage IV and recurrent neuroblastoma at the age of more than 1 year. After intensive consolidative chemotherapy with bone marrow transplantation, 29% of patients lived for 4 years without disease progression.
Surgical treatment Surgical treatment is widely used for radical removal of a localized primary tumor. In addition, in recent years, patients with locally advanced or metastatic neuroblastomas have undergone surgical intervention. After initial chemotherapy, it is often possible to radically remove the primary tumor and metastases due to their significant regression. In some cases, repeated surgical interventions are resorted to if the primary operation turned out to be non-radical, but then the tumor shrank after additional chemotherapy.
Radiation therapy Currently, due to the progressive development of antitumor drug therapy, the role of traditional radiation treatment for patients with neuroblastoma has decreased. This is also due to the fact that this tumor mainly affects young children, whose irradiation carries the risk of long-term radiation damage.
However, in some clinical situations there are indications for its use. Firstly, radiation treatment can be carried out in case of non-radical surgical removal of the primary tumor and low effectiveness of chemotherapy, and secondly, in the presence of a locally advanced inoperable primary or metastatic tumor that does not respond to modern courses of chemotherapy.
The amount of radiation dose primarily depends on the age of the child and the size of the residual tumor. According to Tereb, Tent, when irradiating a child under one year old, an adequate dose may be 10 Gy, delivered over two weeks (1 Gy per fraction). Jacobson (1984), supporting this view, recommends a dose of 12 Gy over two weeks.
In older children, the total dose of ionizing radiation to the tumor should be increased. For example, in children from 1 to 2 years old - up to 15 Gy in two weeks (1.5 Gy daily 5 times a week). At the age of over 3 years, the dose can reach 30-45 Gy, delivered in a single focal dose of 1.5 to 2 Gy 5 times a week (Jacobson, 1984; Scognamillo, 1987, etc.).
Often, after administering the indicated doses, the tumor either completely regresses or significantly decreases in size. In the latter case, surgical removal of the remnants of neuroblastoma becomes possible. A symptomatic effect can be achieved after a single irradiation dose of 5 Gy, but in cases where the child’s condition is relatively satisfactory, it is better to use a course of irradiation with a single dose of 3 Gy, delivered in 5 fractions.
During radiation treatment, it is necessary to capture the entire tumor or its bed with 2 cm of adjacent tissue.
We believe that the issue of using radiation therapy for patients with neuroblastoma should be decided in each case individually. In this case, it is necessary to take into account the possible direct effect on the growing tumor. For example, even with large unresectable tumors, it may be sufficient to administer a radiation dose of 10-15 Gy over 2-3 weeks (1 Gy daily) to achieve an effect due to their spontaneous maturation.
The method of megavoltage irradiation of patients with neuroblastoma is determined primarily by the localization of the tumor process, the size and extent of the tumor. Almost the majority of neuroblastomas are found in the abdominal and thoracic cavities. This dictates the need to use mainly two-field irradiation - two opposing fields.
For tumors of the posterior mediastinum, using photons or electrons, it is necessary to include the entire tumor mass and the thoracic vertebrae to their entire width in the irradiation zone to prevent spinal deformation. Once the upper limit of radiation tolerance of the spinal cord is reached, its protection is necessary. It must be remembered that tolerance changes with age. Careful protection of the shoulder joints is also necessary. For neuroblastomas, irradiation of the entire abdominal cavity is currently rarely used, since aggressive polychemotherapy is usually used in patients with stage III disease.
When irradiating the pelvic area, close attention is paid to protecting the hip joints. In girls, it is necessary, if possible, to remove the ovaries from the zone of action of the direct beam of ionizing radiation by promptly moving them. The latter protects the child from sterilization and preserves its hormonal function.
In recent years, internal irradiation with metaiodobenzylguanidine (mJBG), labeled with radioactive iodine-131, which selectively accumulates in neuroblastoma, has been proposed for the treatment of children with neuroblastoma. The first results of this type of radiation therapy have been published. It was shown that 50% of patients with stage IV disease with a chemoresistant form of the tumor responded positively to the introduction of mJBG (J -131). Mastrangelo et al reported a 10-month-old child with stage III neuroblastoma who was alive without evidence of disease 18 months after diagnosis and treated with internal radiation alone.
Some treatment regimens for children with neuroblastoma depending on the stage of the disease Stage I. Performing radical surgical removal of the tumor is sufficient to cure the patient. However, these children require dynamic monitoring, as relapses of the disease or distant metastases are possible.
Stage IIA . At this stage of neuroblastoma, cure of the patient, as well as at stage I, can be achieved by surgical removal of the tumor.
Two studies showed that 100% of children with and without postoperative treatment were free of recurrence (Ninane et al., 1982; Hayes et al., 1983). The exception was the “dumbbell-like” form of paravertebral neuroblastoma, which requires adequate chemoradiotherapy.
Stage IIB . At this stage of the disease, treatment should begin with chemotherapy followed by surgery. It is difficult to say unambiguously whether there is a need for its use in children under 6 months, since the course of the disease at this age is reliably favorable.
Stage III (the tumor is unresectable) and stage IV . Treatment of children with these stages of the disease is a very difficult task and is solved mainly based on the experience of a specific specialized pediatric oncology department. However, two basic principles for the treatment approach can be pointed out: 1. Aggressive tactics of surgical treatment are necessary for stage III of the disease. 2. Children with stage IV over the age of 1 year require intensive high-dose chemotherapy with a bone marrow transplant.
Stage IVa . Basically, at this stage of the disease, chemotherapy is performed (several courses of cyclophosphamide and vincristine), before and after surgery performed to remove the primary tumor.
Example of polychemotherapy blocks used in modern protocols
PFV/VDIA (GPO, Germany, NB-90) PEV - Cisplatin 40 mg/m2 on day 1 - 4 VP-16 125 mg/m2 on day 1 - 4 Vindesine 3 mg/m2 on day 1 th day. VDIA - Vincristine 1.5 mg/m2 on days 1 and 8 DTIC - 200 mg/m2 on days 1 - 5 Ifosfamide 1.5 g/m2 on days 1 - 5 Doxorubicin on 30 mg/m2 on the 6th, 7th day. Interval 3 weeks.
OPEC/OJEC (UKCCSG, UK) OPEC - Vincristine 1.5 mg/m2 on day 1 Cisplatin 80 mg/m2 on day 1 (infusion 24 hours) Cyclophosphamide 600 mg/m2 on day 1 VP-16 200 mg/m2 on day 2 (infusion 4 hours) In OJEC, cisplatin is replaced by carboplatin 500 mg/m2
CAV-Pt (protocol N 3891, CCG, USA) Cisplatin 60 mg/m2 on day 1, VP-16 100 mg/m2 on days 3, 6. Cyclophosphamide 900 mg/m2 on the 4th, 5th day Doxorubicin 30 mg/m2 on the 3rd day. Interval 3 weeks
Prognostic factors These factors were obtained as a result of identifying a significant relationship between the life expectancy of patients after treatment with age, stage and biological parameters of the tumor. Using multivariate analysis, Evans et al. (1987) determined that a combination of such features as age, stage, serum ferritin level and morphological structure of neuroblastomas allows us to distinguish three groups of children with different prognosis: - favorable, in which more than 80% of patients live for more than 2 years; - intermediate; - unfavorable, in which only about 20% of children are observed for 2 years.
Age at diagnosis is the only important prognostic factor. Thus, in children under 1 year of age the course of the disease is always favorable. Further, significant features that influence the prognosis are the stage and primary location of neuroblastoma.
The prognosis for patients with stages I and IVS, determined according to the international INSS classification, is significantly more favorable compared to children with other stages of the disease. Thus, with stages I and IVS of neuroblastoma, 90% and more than 80% of children, respectively, live for more than 5 years after treatment, while with stages IIA and B, III and IV this figure is 70-80%, 40-70% and 60% are under 1 year of age, 20% are over 1 year of age, and 10% are 2 or more years of age.
At the same time, I would like to note that 80% of children are admitted to a specialized clinic at stages III-IV of the disease.
Retroperitoneal and, in particular, adrenal neuroblastoma has a worse prognosis, and a tumor localized in the mediastinum has the best prognosis compared to other tumor localizations. Morphological signs of differentiation in the primary tumor are a prognostically favorable factor, as are hyperdiploid type blastomas.