About the disease
Multiple system (multifocal, multisystem) atrophy is the extensive degeneration of glial cells in all structures of the brain (BM). The disease is abbreviated as MSA or MSA. Multiple system atrophy is a rapidly progressive, severe disease in which the spinal cord is often involved in the pathological process, leading to a decrease in the tissue of the central nervous system and disability.
Degeneration provokes the appearance of:
- dementia;
- shaking palsy - parkinsonism syndrome;
- dysfunction of the autonomic nervous system;
- cerebellar ataxia - a disorder of motor and speech function;
- insufficiency of the pyramids - the functioning of the central nervous system is disrupted.
Reference! MSA is a rare incurable pathology that occurs predominantly in men. The incidence is less than 6 cases per 100,000 population.
During the development of the disease, the subcortical nuclei (basal ganglia) inside the brain hemispheres are affected. They are located between the diencephalon and the frontal lobes. They consist of the striatum, gray and white matter, nigral (black) substance, and subthalamic nucleus. Histological examination of oligodendrogliocytes reveals accumulations of alpha-synuclein in cytoplasmic inclusions. This is unusual for the composition of neuroglial cells.
As a result of degeneration of the subcortical nuclei, the following is disrupted:
- coordination of movements;
- regulation of muscle tone;
- sensitivity of perception of visual, auditory and other stimuli;
- regulation of trophism, metabolism, respiration, urination, and other vegetative functions;
- development of reflexes, memory, other regulation of higher nervous activity.
MSA is more often detected in people over 50 years of age who have worked with harmful and toxic substances, pesticides, formaldehyde, and solvents. Patients with Parkinson's disease and Alzheimer's disease are at risk. The danger is that the pathology causes an irreversible process in the brain with the death of nerve cells. From the moment symptoms appear, life expectancy does not exceed 15 years. Death often occurs due to respiratory failure or sepsis.
Figure 1. Imaging of the brain during tomography.
Until 2021, in ICD-10, multiple system atrophy was classified as code G90.3 under the name “multisystem degeneration.” Now in the directory there remains the parkinsonian type of MSA under the code G23.2 “MSA-P” and the cerebellar type – G23.3 “MSA-C”. In ICD-10-CM, pathology is numbered G90.3.
Magnetic resonance imaging (MRI) in St. Petersburg
MRI of the brain. Demonstration of atrophy and normality in color treatment.
The term “neurodegenerative diseases” (NDDs) defines a large group of diseases, predominantly of late age, which are characterized by slowly progressive death of certain groups of nerve cells and, at the same time, gradually increasing atrophy of the corresponding parts of the brain and/or spinal cord. The development of these diseases is based on metabolic disorders and changes in the conformation of cellular proteins with their subsequent accumulation and aggregation in certain groups of neurons. In NDD, predominantly neurons and glial cells of the basal ganglia and stem structures that produce acetylcholine, dopamine, and serotonin are affected.
The classification divides NDD into 2 large groups – sporadic and irritative.
- Sporadic NDD:
- Progressive supranuclear palsy (Steele-Richardson-Olszewski disease).
- Multiple system atrophy.
- Dementia with Lewy bodies.
- Parkinsonian dementia (Guam syndrome).
- Corticobasal degeneration.
- Alzheimer's disease.
- Irritative NDZ:
- Huntington's disease.
- Hallerwarden-Spatz disease.
- Wilson-Konovalov disease.
- Farah's disease.
- Bessen-Kornzweig disease.
Alzheimer's disease is a progressive neurodegenerative disease characterized by the gradual development of dementia. The origin of the disease is precisely unknown. Biochemical changes consist of a decrease in the activity of choline acetyl transferase in the cerebral cortex and hippocampus. Pathological manifestations include the formation of specific amyloid plaques, neurofibrillary strands and reactive gliosis. Atrophy develops, predominantly affecting the cortex around the Sylvian fissures and the hippocampus, with secondary expansion of the ventricles, especially the temporal horns
The disease was first described by Alois Alzheimer in 1907. The process resembles natural aging, but sharply accelerated. It begins with memory impairment, then loss, inability to perform daily self-care, and repeated questions. Later, profound mental and speech disorders, weight loss, and convulsions appear.
The incidence is 0.51% for individuals aged 70–74 years, with a progressive increase in incidence with age. Clinical manifestations include memory impairment, depression, behavioral disturbances and hallucinations. In the later stages, extrapyramidal symptoms are added to mental disorders. The disease ranks 4th in mortality. Diagnosis is made on the basis of clinical and neurophysiological examination, as well as neuroimaging. Typical CT findings include diffuse atrophy (especially of the temporal lobes), secondary widening of the sulci and ventricles. Sensitivity (without volume measurements) in comparison with the normal age group is about 80%, specificity is about 70%. Measuring hippocampal volumes using thin-slice MRI increases accuracy to 85%.
MRI of the brain. T2-weighted sagittal MRI. Pick's disease. Color processing of the image.
MRI of the brain is the method of choice for assessing structural changes. Atrophic changes are expressed throughout the mediobasal region of the temporal lobe. The sensitivity and specificity of MRI for early dementia is about 80%. Measuring hippocampal and amygdala volumes improves accuracy to 85%.
MRI of the brain. T1-weighted coronal MRI. Diffuse atrophy in Alzheimer's disease.
Differential diagnosis with MRI of the brain should be carried out with Parkinson's disease, multi-infarct dementia and frontoparietal dementia (Pick's disease).
These same areas show hypoperfusion and decreased activation on fMRI. In addition to MRI, [18F]fluoro-2-deoxyglucase (FDG) PET is important in the study of Alzheimer's disease. Hypometabolism correlates well with the severity of the disease and predicts its development.
Parkinson's syndromes include a group of diseases clinically close to Parkinson's disease. Parkinson's syndromes include rapidly progressive dementia with Lewy bodies. In MRI of the brain, a low signal is observed not only from the compact part of the substantia nigra, but also from the putamen, which becomes even darker than the globus pallidus. With olivopontocerebellar atrophy, sagittal MRI of the brain shows a decrease in the volume of the pons and cerebellum. With progressive supranuclear palsy, atrophy of the quadrigeminal plate is detected. The characteristic symptoms of MRI are described - “penguin”, “Mickey Mouse” and others, the meaning of which is to describe the signs of atrophy.
In dementia associated with Parkinson's disease, MRI of the brain shows a decrease in the thickness of the cortex in the projection of the parahippocampal part of the left middle occipitotemporal gyrus and a decrease in the volume of the left inferior longitudinal fasciculus. Decreased left parahippocampal cortical thickness is associated with a high risk of depression. It was noted that daytime sleepiness correlates with a decrease in the thickness of the fusiform cortex, determined by MRI.
To track dynamics and forecast, various measurements are also used with MRI of the brain:
- The average brain-pontine coefficient is normally 0.24, and with progressive supranuclear palsy it becomes less than 0.12.
- Parkinsonism index - the ratio of the width of the superior cerebellar peduncle in the coronal plane to the area of the midbrain in the midsagittal plane multiplied by the ratio of the width of the middle cerebellar peduncle to the width of the superior cerebellar peduncle - more than 13.55 indicates in favor of parkinsonian syndromes. MRI reveals atrophy of the caudate nuclei with secondary expansion of the anterior horns; atrophy of the putamen and cortex of the frontal lobes. The ratio of the width of the anterior horns to the distance between the caudate nuclei (along their edges), measured in the transverse plane, decreases from 2.2-2.6 to values close to 1.0. Another coefficient - the distance between the caudate nuclei (between their heads to the width of the skull along the internal plates) - increases (normally 0.09-0.12). MRI of the brain reveals diffuse brain atrophy, expansion of the Virchow-Robin perivascular spaces and leukoaraiosis. The latter is a consequence of stenosis and occlusion of the deep veins of the brain. On T2-dependent MRI images, leukoaraiasis appears as small foci of hyperintensity. In general, these signs are nonspecific and reflect aging of the brain. In MRI of the brain, tomograms of both types of weighting reveal an increased signal from the pons and tegmentum of the cerebellum. A typical signal change is from the periphery of the bridge. Unlike tumors, there is no swelling or mass effect on MRI. The earliest manifestations are detected on diffusion-weighted MRI of the brain, approximately 24 hours after the onset of tetraparesis.
Progressive supranuclear palsy manifests as disturbances in upward gaze, extrapyramidal symptoms, and mental impairment. The disease develops in people around 60 years of age. The etiology is unknown, almost all cases are sporadic. The frequency is 1-1.5 cases per 100 thousand population. The disease is characterized by an abnormal accumulation of tau protein in the brain. MRI of the brain shows diffuse atrophy, with a characteristic “penguin” sign noted on sagittal T1-weighted MRI. Atrophic changes in the pons and midbrain lead to expansion of the aqueduct and the third ventricle, the contour of which resembles the outline of a penguin.
MRI of the brain. T12-weighted sagittal MRI. Progressive supranuclear palsy. Penguin symptom.
Central pontine myelinosis (osmotic dementia) is an acquired metabolic disorder. More common in alcoholics. Hyponatremia results in demyelination visible on MRI. Central pontine myelinosis is often accompanied by extracentral myelinosis, when there is a lesion above the trunk. Clinical manifestations are limited to lethargy (even lethargy), spastic tetraparesis and damage to the lower cranial nerves.
MRI of the brain. T2-weighted axial MRI. Central pontine myelinosis,
Binswanger's disease (subcortical atherosclerotic encephalopathy, small vessel dementia). This is a condition associated with multiple infarctions of small branches, which are visible on MRI of the brain as lacunar strokes. The disease gradually progresses. A variant of Binswanger's disease can be considered an inherited familial arteriopathic leukoencephalopathy.
MRI of the brain. T2-weighted FLAIR MRI. Binswanger's disease.
Huntington's disease is an inherited disease that appears in middle age and progresses rapidly. The clinical picture is dominated by choreoathetosis and dementia.
MRI of the brain. T1-weighted coronal MRI. Huntington's disease.
Fahr's disease is a very rare hereditary disease manifested by calcification of the basal ganglia and dentate nucleus. On T2-weighted MRI, the nuclei are sharply hypointense, consistent with calcifications clearly visible on CT. Often, small hyperintense foci are found in the area of the visual tuberosities.
MRI and CT scan of the brain in the axial plane. Farah's disease.
You can also read about MRI in St. Petersburg for neurodegenerative diseases on the page of our other website. We perform the study in a closed machine, but it is also possible using an open MRI. MRI St. Petersburg allows you to choose the location of the MRI, but in this case we recommend that you be examined by us.
Leave feedback.
MRI in St. Petersburg USA
Reasons for development
Scientists continue to study the causes, mechanism of development and triggering factors of MSA. There is no clear confirmation of a hereditary predisposition to the development of the disease. In a child, congenital atrophy of the brain occurs when a woman abuses medications, alcohol, or drugs during pregnancy.
During diagnosis, doctors identify possible causes of multisystem atrophy:
- Parkinson's or Alzheimer's disease;
- contact with neurotoxic substances;
- variability of the α-synuclein gene;
- alcohol and drug poisoning in people with addiction;
- brain and/or spinal cord injury;
- hypoxia of brain tissue.
Pathogenesis is difficult to study because the exact causes of multiple system atrophy are unknown. Studies have revealed the accumulation of tau protein in affected oligodendrogliocytes. This is detected in the cerebellum, pyramids, brain cortex, horns of the spinal cord in the thoracic and sacral regions of the ridge. At the same time, dopamine receptors and the substantia nigra are damaged, and α-synuclein accumulates in neuroglial cells.
Reference! MSA is characterized by an asymmetric decrease in white matter and impaired transmission of nerve impulses. Neurons suffer less than oligodendrogliocytes.
Classification
Doctors distinguish three forms of MSA depending on which syndrome is identified by the presenter. If this cannot be established, the patient is diagnosed with a mixed type of disease.
Classification of pathology:
| Leading symptom | Type of multiple system atrophy | Distinctive features |
| Parkinsonism | Striatonigral type of MSA | Slow movements, mask-like face, freezing in one position, “air cushion” symptom, tremors of the limbs, bent joints, decreased mobility. The striatum, the substantia nigra, is more susceptible to degeneration. |
| Autonomic failure | Shy-Drager syndrome | Dysfunction of glands and organs, pelvic disorders, hypotension, snoring, apnea. |
| Cerebellar ataxia | Olivopontocerebellar type of MSA | Poor balance, impaired fine motor skills, involuntary movement of the eyeballs, muscle weakness. The cerebellum, pons, and olives are more susceptible to degeneration. |
In the classifiers, doctors propose to remove Shy-Drager syndrome, since autonomic failure accompanies all forms of MSA. ICD-10 lists only the cerebellar and parkinsonian types of the disease.
Clinical varieties
Depending on the combination and prevalence of symptoms, several clinical types of multiple system atrophy are distinguished:
- Strionic form with dominant parkinsonism. Its manifestations mask other, less pronounced symptoms. Unlike Parkinson's disease, there is usually no period of hemiparkinsonism with one-sided tremor and rigidity. Trembling can be combined with myoclonic twitching of small muscles, acquiring an arrhythmic and uneven character. Low sensitivity to dopamine-containing drugs is characteristic, and the initial effects of treatment quickly fade away.
- Olivopontocerebellar form, with a predominance of cerebellar disorders.
- Vegetative form, or Shy-Drager syndrome. Severe and poorly corrected orthostatic hypotension becomes the main factor of disability, leading to social and everyday disadaptation already in the early stages of the disease. It is diagnosed if, in an upright position, systolic pressure decreases by 20 or more mmHg, and diastolic pressure by 10 or more mmHg. (compared to the level in the supine position). Characteristic complaints are fatigue, non-systemic dizziness, presyncope and syncope, acrocyanosis. Orthostatic hypotension can be supplemented by nocturnal arterial hypertension and low pulse adaptability to stress.
Most often, a combination of parkinsonism with autonomic disorders occurs, and as symptoms develop, the type of disease may change. The addition of cerebellar disorders aggravates the condition, having the greatest impact on the ability to move independently safely.
Symptoms
The first sign of MSA is the onset of disease progression at an older age after 45 years. Symptoms develop quickly. Most people immediately develop parkinsonism and movement disorders. In 40% of cases, degeneration starts with autonomic dysfunction.
Changes in the initial stage are not always noticed by the patient. Among the first signs are pelvic disorders: erectile dysfunction, difficulty urinating or defecating, urinary/fecal incontinence. Every fifth patient begins to fall from the moment of progression of multiple system atrophy. The cause is considered to be orthostatic hypotension, muscle weakness, and cerebellar dysfunction.
Reference! The progression of MSA is characterized by the addition of other symptom complexes to the leading syndrome. That is, a person simultaneously exhibits autonomic failure in combination with signs of parkinsonism and cerebellar ataxia.
Differences in symptoms between different types of MSA
Primary symptoms depend on the class of multiple system atrophy. With the striatonigral type of MSA, signs of Parkinson's disease are immediately noticeable. Initially, the body responds to treatment with levodopa, then the effectiveness of the drugs is lost, and autonomic disorders worsen.
Primary signs of parkinsonian type of multiple system atrophy:
| Symptom | Explanation |
| Bradykinesia | All voluntary movements slow down. A person walks, speaks, writes, reads aloud more slowly. Coordination of movements and speech is preserved. A long conversation or the need to move causes rapid fatigue. |
| Rigidity | Stiffness of movement, tension in the muscles responsible for contraction and extension. The chin almost touches the clavicular area. In a horizontal position on the back, the head does not lie on the pillow, but this disappears after falling asleep. The limbs are bent at large joints, the torso is bent forward, the spine is stooped. During passive movement of the limb (performed by the doctor) under the fingers, the doctor feels the viscous resistance of the muscles. |
| Postural instability | A person cannot maintain balance. This is not associated with orthostatic hypotension, darkening of the eyes, or hypertension. |
| Tremor | The muscles of the torso, neck, arms, legs tremble during movement or rest. The tremor disappears when the patient performs the opposite action. That is, it stops or starts to move. |
With the olivopontocerebellar type of MSA, symptoms of cerebellar dysfunction are in the foreground. The patient begins to mince (step length decreases). There is unsteadiness of gait, muscle stiffness, deterioration in overall coordination of movements and fine motor skills. The tremor intensifies when approaching the target of the movement. It is difficult to change rapidly alternating actions.
With the cerebellar type of multisystem atrophy, dysarthria and oculomotor (oculomotor) dysfunction are manifested. Their symptoms:
- Muffled voice;
- Extended pronunciation of words;
- Chanted speech;
- Violation of sound modulation, phonation, breathing during pronunciation;
- Rhythmic involuntary movement of the eyeballs (nystagmus).
Shy-Drager syndrome in MSA is manifested by a disorder of the functions of the pelvic organs and glands. Fainting or collapse occurs due to a drop in pressure. Signs include impaired urination, bowel movements, decreased salivation, lacrimation, and sweating. Eye movements, conversation, and short-term cessation of breathing are observed during sleep. In men, erection deteriorates and impotence develops.
Reference! The progression of MSA is manifested by worsening symptoms of types 1–3 of multiple system atrophy. The clinic is supplemented by dementia, paralysis or paresis, inappropriate behavior, and complications of degeneration.
International Neurological Journal 5 (51) 2012
Parkinsonism is a progressive disease of the nervous system, characterized by slowness of voluntary movements, muscle rigidity, trembling (tremor) at rest, poor facial expressions, changes in gait (small steps, lack of normal swinging of the arms). The most common form of parkinsonism is idiopathic parkinsonism (or Parkinson's disease), the main distinguishing manifestations of which are rigidity, resting tremor, impaired postural reflexes, asymmetry of onset, good response to levodopa, cell death of the substantia nigra and the presence of Lewy bodies [1, 2]. But there are several other primary neurodegenerative diseases with damage to the extrapyramidal system, in which parkinsonism syndrome (Table 1) may be one of the main or additional manifestations [5, 7, 8].
In the early stages of the disease, there are difficulties in differential diagnosis of various forms of parkinsonism. The diagnosis of various parkinsonian disorders is significantly improved by the use of special criteria, such as the system for assessing the severity of parkinsonism by L.S. Petelin et al. (1980), the Hoehn-Yahr scale (1967) as modified by Lindval et al. (1989), Tetrud, Langstone (1989), Unified Parkinson's Disease Rating Scale (Fahn, Elton, 1987), UK PD Society diagnostic criteria for Parkinson's disease (Gibb, Lees, 1988), diagnostic criteria for multiple system atrophy (Gilman et al., 1998 ), criteria for the diagnosis of progressive supranuclear palsy (NINDSSPSR criteria Litvan et al., 1996), clinical neuroimaging criteria for the diagnosis of vascular parkinsonism (Levin O.S., 1997), criteria for the diagnosis of essential tremor (Elble, 2000), criteria for the diagnosis of Tourette's syndrome (The Tourette Syndrome Classification Study Group, 1993) and others [8–10].
Using a combination of criteria on different scales allowed us, already at the clinical stage, to carry out a differential diagnosis of the etiological factor of parkinsonism syndrome, which we observed in a 60-year-old patient.
In August 2011, to the Department of Angioneurology and Neurorehabilitation of the State Institution “Institute of Emergency and Reconstructive Surgery named after. VC. Gusak NAMS of Ukraine" patient R., 60 years old, was admitted with complaints of stiffness in the arms and legs, weakness in them, unsteadiness when walking, slow gait, clumsiness, slow speech, urinary incontinence, darkening in the eyes when changing body position, a tendency to falls, decreased memory for recent events.
He has been ill for about 10 years, when he began to notice instability when walking and weakness in his legs. I didn’t attach any importance to this. The disease progressed slowly. Over the past 3 years, he has noted a significant deterioration in his condition: severe stiffness in the legs, difficulty walking, unsteadiness when walking. I was seen by a neurologist for men and women with a diagnosis of dyscirculatory encephalopathy (atherosclerotic) stage 1–2. with parkinsonian syndrome, akinetic-rigid form, progressive course with reflex-pyramidal insufficiency in the extremities. I took mirapex, levokom, nacom. There was practically no effect from the therapy. Hereditary history is not burdened. He was sent to the department due to the lack of effect of treatment and the appearance of autonomic disorders (sweating, low blood pressure, weakness, dizziness, urinary incontinence, syncope, i.e. symptoms of pandysautonomia).
Objectively: the condition is relatively satisfactory. Increased greasiness of the facial skin with the presence of hyperemia in the area of the nasolabial triangle, forehead and eyebrows, peeling of the skin. Marble pattern of the skin of the feet. Peripheral lymph nodes are not enlarged. In the lungs there is vesicular breathing, no wheezing. Muffled heart sounds. Blood pressure 110/70 mm Hg. Heart rhythm - 64 per minute. The abdomen is soft, sensitive along the large intestine, in the area of the white line there is a hernial protrusion when straining. There is no swelling. Pasternatsky's symptom is negative.
Neurological status: hypomimia, bradykinesia, bradyllalia. Muscle tone is increased according to the extrapyramidal type - viscous, plastic, monotonous, intensifies with each repeated passive movement (the “wax doll” phenomenon), Neuck’s symptom on both sides. Postural tremor of the limbs, postural instability. Acheirokinesis. Palpebral fissures D < S, pupils are equal, reactions to light are lively. Weakness of abduction in all directions, convergence. Horizontal nystagmus in extreme abductions. The soft palate is inactive. Palatal reflexes are not evoked. Tongue in the midline. Dysphonia. Dysarthria. Positive reflexes of oral automatism. Hand reflexes are high S > D. Knees are high D > S, Achilles are high D > S. Abdominal reflexes are decreased. Pathological wrist reflexes. Deep pain sensitivity is not impaired. Vibration sensitivity at ankles D - 12 s, S - 14 s. Coordination tests are performed with dysmetria and intention tremor. Babinsky asynergy, Stewart-Holmes symptom. The gait is shuffling, with legs widely spaced.
A study of the function of the autonomic nervous system revealed:
- cardiovascular disorders in the form of orthostatic hypotension (80/50 mm Hg), arterial hypotension after meals (90/60 mm Hg), arterial hypertension in the supine position, decreased heart rate variability;
- gastrointestinal disorders in the form of complaints of discomfort in the epigastrium after eating, hypersalivation, nausea, constipation and a feeling of incomplete bowel movement during defecation;
- genitourinary disorders in the form of urinary disorders of the irritative type, impaired potency;
- trophic disorders in the form of dryness, thinning of the skin and the presence of seborrheic dermatitis;
- violation of pupillary innervation in the form of complaints of blurred vision in dark or brightly lit rooms;
- livedo reticularis on the skin of the feet.
Additional examinations: general clinical and biochemical tests of blood, urine and cerebrospinal fluid, electrolytes, blood sugar without pathology. Blood cholesterol - 4.7 mmol/l, Reactive protein - 2 mg/l, rheumatoid factor - 8 IU/ml, ASLO - 54 U/ml, blood ceruloplasmin - 0.25 g/l, copper in daily urine - 28 mcg /day, adrenaline in the urine - 14 mcg/day, norepinephrine in the urine - 45 mcg/day, dopamine in the urine - 438 mcg/day. Total protein - 75 g/l, albumins - 43 g/l, a1globulins - 5.8%, a2globulins - 8.7%, bglobulins - 10.1%, gammaglobulins - 16.4%. Liquor pressure was 140 mm water column.
MRI of the brain - high signal intensity in T2 mode in the area of the pons and middle cerebellar peduncles reflects degeneration of pontocerebellar fibers, a decrease in signal intensity in the putamen area. Reduced cortical volume in the primary sensorimotor, lateral premotor and prefrontal areas, decreased cerebellar volume.
MRI of the cervical spine and spinal cord 09/14/11 - osteochondrosis of the cervical spine with protrusions of the C4C5, C5C6, C6C7 discs. Spondylosis deformans, spondyloarthrosis. No additional formations or foci of pathologically altered MR signal were identified in the spinal cord and extramedullary spaces.
Duplex scanning of the main arteries of the head - the lumen of the CCA, the ICA is clean, no intraluminal formations were detected, differentiation of the arterial wall into layers is preserved, IMT is 1.1 mm, moderate hemodynamically insignificant S-shaped tortuosity of the ICA on both sides.
ECG - sinus rhythm, vertical position of the EOS, heart rate 78 per minute.
Urologist - neurogenic bladder.
The MMSE scale and the clock drawing test were used to assess the degree of cognitive impairment. The results obtained were 28 and 10 points, respectively.
Differential diagnosis was carried out between Parkinson's disease, parkinsonism "plus", progressive supranuclear palsy, corticobasal degeneration, normal pressure hydrocephalus, secondary dysmetabolic encephalopathies, and multiple system atrophy.
Based on the clinical picture, the nature of the course of the disease, data from additional studies, and the above diagnostic criteria, we came to the conclusion that the patient has multisystem brain atrophy with severe akinetic-rigid and ataxic syndrome, reflex-pyramidal insufficiency in the limbs, autonomic failure (pandysautonomia), and severe impairments of movement and self-care.
Received treatment: Levocom 600 mg/day in 3 doses, neomidantan 100 mg orally 2 times a day, Cortexin 2.0 IM, Cytoflavin 10.0 IV drip at 200.0 physiological solution No. 7, then orally 1 tablet 2 times a day inject bellataminal 1 tablet 3 times a day, kudesan Q10 10 drops 2 times a day with meals, Semax 0.1% solution 2 drops into each nasal passage 2 times a day.
Background: multisystem atrophy is a sporadic progressive neurodegenerative disease affecting the basal ganglia, brain stem, cerebellum, and spinal cord, manifested by parkinsonism, cerebellar ataxia, autonomic failure, and pyramidal syndrome in various combinations. Multiple system atrophy is an independent nosological form, which is one of the variants of multisystem degenerations.
Depending on the predominance of certain syndromes, there are 3 main clinical types of MSA:
1) striatonigral degeneration (striatonigral type), characterized by a predominance of parkinsonian symptoms in the clinical picture;
2) olivopontocerebellar atrophy (olivopontocerebellar type), characterized by the predominance of cerebellar ataxia in the clinical picture;
3) Shy-Drager syndrome, characterized by the dominance in the clinical picture of symptoms of progressive autonomic failure, primarily orthostatic hypotension.
Diagnosis criteria for multiple system atrophy (Gilman et al., 1998)
Clinical manifestations (characteristic of the disease):
Autonomic/pelvic dysfunction (orthostatic hypotension with a decrease in systolic pressure by at least 20 mm Hg or diastolic pressure by at least 10 mm Hg within 3 minutes of standing; urinary incontinence and incomplete emptying of the bladder).
Parkinsonism (hypokinesia; rigidity; postural instability not associated with primary impairment of vision, proprioception, vestibular or cerebellar functions; resting tremor and/or postural tremor).
Cerebellar ataxia (statolocomotor ataxia with an increase in the area of support, uneven steps in length and direction; scanned speech; incoordination of the limbs; nystagmus).
Pyramid syndrome (revitalization of tendon reflexes with the presence of extensor foot signs).
Criteria for diagnosis:
Autonomic/pelvic dysfunction (orthostatic hypotension with a decrease in systolic pressure by at least 30 mm Hg, diastolic pressure by at least 15 mm Hg after 3 minutes of standing and/or urinary incontinence and erectile dysfunction in men).
Parkinsonism (hypokinesia in combination with at least one other parkinsonian symptom).
Cerebellar ataxia (statolocomotor ataxia in combination with at least one other cerebellar symptom).
Criteria excluding diagnosis:
Onset before age 30; positive family history; the presence of anamnestic, clinical or paraclinical signs of another disease that can cause similar symptoms; hallucinations not related to taking medications; presence of dementia; a sharp slowdown in vertical saccades or vertical gaze paralysis; signs of dysfunction of cortical functions (aphasia, alien hand syndrome, dysfunction of the parietal cortex).
Currently, there is no effective treatment for cerebellar disorders in MSA, so pharmacotherapy is largely aimed at alleviating the symptoms of parkinsonism and dysautonomia. Levodopa (plus DOPA decarboxylase inhibitor) at a dose of up to 1000 mg/day if well tolerated. Dopamine receptor agonists are second-line drugs (doses are the same as for Parkinson's disease), amantadine is a third-line drug (100 mg up to 3 times a day), Semax 0.1% solution nasal drops. Therapy for orthostatic hypotension is often quite complex, but it improves the quality of life of patients with MSA. Low blood pressure may not be accompanied by any symptoms, probably due to the fact that their cerebral blood flow is maintained at an adequate level due to autoregulatory mechanisms even when systolic blood pressure decreases to 60 mmHg. When this condition creates discomfort, it can be avoided by limiting the action of provoking factors - large amounts of food, alcohol, physical activity, external heat exposure. Non-pharmacologic strategies include wearing elastic tights, elevating the head of the bed at night, and increasing the salt content of the diet. Urological problems in MSA are caused by a combination of central and peripheral neurological disorders, which are sometimes superimposed on local pathological changes, such as prostate hypertrophy and weakness of the perineal muscles. Peripheral anticholinergics are effective for urinary incontinence, but often induce urinary retention; Desmopressin taken at night provides regression of nocturia. If the bladder is not completely emptied, periodic self-catheterization is necessary. Because the results of pharmacotherapy for MSA are generally poor, the role of other therapeutic strategies is important. Physiotherapy helps maintain mobility and prevent contractures, speech therapy improves articulation, communication and swallowing, occupational therapy helps overcome the limitations of permanent disability, and psychotherapy provides emotional support for both the patient and family. Dysphagia is often accompanied by the need for feeding through a nasogastric tube and even the use of percutaneous endoscopic gastrostomy. Providing the patient with a wheelchair is necessary due to the tendency to frequent falls and gait ataxia.
Currently, the understanding of the clinical manifestations of MSA has improved [6–8]. The same cannot be said for treatments whose results are moderate or non-existent. Therefore, there is an urgent need to develop future therapeutic studies aimed at exploring new symptomatic and neuroprotective drugs, as well as optimizing non-pharmacological interventions for this pathology.
Diagnostic methods
You need to be examined by a neurologist. To make a diagnosis, dynamic observation of the patient using cerebral MRI is necessary. In case of contraindications, computed tomography PET and SPECT are performed.
At the beginning of the development of MSA, MRI will not show atrophic changes in brain tissue, but will help to exclude a tumor, encephalitis, and multiple sclerosis. After 1–3 years of intensive progression, expansion of the fourth ventricle, pronounced degeneration of the subcortical ganglia, the lower half of the pons, the cerebellum, and the putamen are revealed.
During the examination, the neurologist assesses the presence of autonomic failure in combination with parkinsonism and/or cerebellar dysfunction.
Multiple system atrophy is not confirmed if:
- MSA began to develop before age 30 or after age 75;
- autonomic failure is not combined with either cerebellar dysfunction or parkinsonism;
- close relatives also have pathology (family history);
- The patient has dementia and signs of a disease similar to MSA;
- Treatment of parkinsonism is effective with levodopa medications.
To make a diagnosis, an orthostatic test examines the functions of the autonomic nervous system. Disruption of the pelvic structures is detected by electromyography of the sphincters.
The development of multiple system atrophy is indicated by the presence of:
- orthostatic hypotension - a decrease in pressure after taking a vertical position;
- irregular tremors;
- coldness of the feet and hands, stiffness of their joints;
- snoring that has reappeared or intensified;
- crying or laughter that is inappropriate to the emotion being experienced;
- severe speech and voice disorders - dysarthria, dysphonia;
- difficulty breathing - inspiratory shortness of breath;
- urinary incontinence;
- erectile dysfunction;
- torticollis with a tilt of the head to the chest - anterocollis;
- involuntary movements of facial muscles, tongue - orofacial dystonia;
- curvature of the spine in the thoracolumbar junction with the torso tilted forward - camptocormia;
- increased incidence of falls;
- cerebellar syndrome/parkinsonism + autonomic failure.
Reference! MSA is reliably confirmed by pathomorphological examination of neuroglia. During life, biomaterial with glial cells is obtained through a brain biopsy or tissue is removed by a pathologist during an autopsy.
Parkinsonism: clinical picture, diagnosis and differential diagnosis
MMA named after I.M.
Sechenov Clinical picture
Parkinsonism (P) is a syndrome associated with damage to the basal ganglia and their connections. Its main manifestations are lack of movement (akinesia) and increased muscle tone (rigidity). Therefore, P is also called akinetic-rigid syndrome.
.
In its literal sense, the term “akinesia”
means the absence of movements or their impoverishment. Akinesia is manifested by the absence of so-called associated, spontaneous movements - for example, during walking there is no physiological synkinesis in the form of swinging arms or the amplitude of this movement is sharply reduced (acheirokinesis); facial expressions are impoverished - rare blinking is noted, emotional movements are reduced (hypomimia). The concept of akinesia also includes difficulty in starting (initiating) movements. With P, there is also a slowness of voluntary motor acts and their exhaustion, which some researchers call the term “bradykinesia”. For example, when repeatedly squeezing and unclenching the hand, pronation-supination of the arms, tapping the foot on the floor, etc., it is noticeable that the amplitude and strength of the first movements is greater than the subsequent ones. In addition to akinesia, impoverishment of movements is designated by the same term - “hypokinesia” (and syndrome P - hypokinetic-rigid syndrome).
Rigidity
- This is a special type of increase in muscle tone. During passive movements in the joints (flexion-extension, pronation-supination, etc.), the researcher feels constant resistance, the same throughout the entire movement. This sensation is similar to that which occurs when bending and extending a lead pipe (in contrast to another type of increased tone - spasticity, in which the greatest resistance to passive movements is noted at the beginning of the movement. The latter is called the “jackknife phenomenon”). Rigidity in P is determined both in the muscles of the neck and torso, and in the muscles of the limbs. Often, an increase in tone predominates in the flexor muscles, which results in a bent posture of the body with the arms slightly bent at the elbow joints and brought to the body and the legs bent at the knee joints. This position is called the “flexor pose” or “supplicant pose.”
Another characteristic symptom is the "gear wheel"
" It occurs along with rigidity, but can also exist in its absence. During flexion and extension of the joints, the researcher feels uneven resistance to the movement produced, creating the impression of sliding gear teeth interlocking with each other.
At the onset of P, many patients do not notice any abnormalities. Often the patient's relatives and friends first notice decreased facial expression (often interpreting this as a sign of depression), acheirokinesis, and slowness of movement, especially when dressing, eating, and walking. Subsequently, the patients themselves note a decrease in manual dexterity, especially when performing fine movements, such as fastening buttons and tying shoelaces. Habitual activities that require manual dexterity (for example, playing a musical instrument, working on a typewriter or using a computer) are disrupted. Writing disorders occur - handwriting changes, becomes small (micrography), less legible, and at advanced stages completely incomprehensible; difficulties brushing teeth, shaving, etc. Turning in bed is significantly difficult, getting up from a chair or low chair is a problem, patients are often unable to get into and out of the bath, so they can only wash in the shower. Many patients associate these phenomena with “weakness” and make corresponding complaints. Lack of energy and fatigue are also common complaints. Subsequently, eating becomes difficult due to difficulty chewing, and choking occurs when swallowing. Due to akinesia of the pharyngeal muscles, swallowing movements become less frequent, which causes the development of drooling. The latter can be so intense that patients are forced to constantly use a scarf or towel.
Often akinesia and rigidity are combined with tremor
. A typical parkinsonian tremor is very unique and difficult to confuse with another type of tremor. Firstly, in the vast majority of cases it is noted in the arms, less often in the legs. It should be remembered that parkinsonian tremor never begins in the head, and even in very advanced stages of P, head tremor is extremely rare. Trembling of the lower jaw and tongue, on the contrary, can be observed quite often (usually in the later stages). Secondly, parkinsonian tremor is a resting tremor. This means that it is observed (or is most pronounced) in a situation when the patient’s hands lie in a relaxed state on his knees, arms of a chair, table, etc. When making movements, the trembling disappears or becomes much less intense. Thirdly, with parkinsonian tremors, the patient’s hands make circular movements, while the thumb and index fingers move towards each other. This is reminiscent of the movements made when counting money or twisting paper balls. Therefore, parkinsonian tremor is figuratively called a tremor of the “counting coins” or “rolling pills” type. It should be remembered that trembling is not an obligatory component of P. syndrome.
Parkinson's disease. Figure A shows a section of the brain of a healthy person with well-pigmented substantia nigra. Figure B shows a section of the brain of a patient suffering from Parkinson's disease. There is a noticeable lack of pigmentation of the substantia nigra.
P is also accompanied
by imbalance due to a disorder of
the so-called postural reflexes.
The latter ensure uprightness, maintaining stability during various movements (hand movements, walking, sitting down and standing up, bending, etc.), as well as during sudden destabilizing influences (for example, when pushing, stumbling, etc.). Disorder of postural reflexes manifests itself in frequent falls of patients. With milder disorders, falls are not observed, but instability occurs when a slight push to the chest or back occurs. When sitting down on a chair, patients fall onto it, often abruptly leaning back. Postural disturbances also cause the occurrence of propulsions -
one of the characteristic features of P. Propulsions are expressed in the fact that when walking the patient is “pulled forward”, he quickly walks forward in short steps, as if “catching up with his center of gravity”, and is not able to stop abruptly. Often this ends in a fall.
P is a syndrome that can occur in various diseases. The most common cause of P is idiopathic parkinsonism, or Parkinson's disease (PD).
.
P is often observed in other idiopathic degenerative diseases of the nervous system. The latter are often called parkinsonism-plus. This group includes multiple system atrophy, progressive supranuclear palsy, diffuse Lewy body disease, and corticobasal degeneration. Symptomatic P (not associated with a primary degenerative disease of the nervous system)
often occurs .
It includes medicinal, vascular, post-traumatic, post-encephalitic, toxic P, P for brain tumors and hydrocephalus. P also characterizes the clinical picture of some hereditary degenerative diseases of the central nervous system
. These include hepatolenticular degeneration (Wilson-Konovalov disease), spinocerebellar ataxia, familial calcification of the basal ganglia, Huntington's disease, etc. Thus, syndrome P is not synonymous with PD.
Diagnosis and differential diagnosis
Essential tremor
At the first stage of diagnostic work, it is necessary to make sure that the patient really has syndrome P. Most often, essential tremor
(ET) and peculiar gait changes associated with vascular pathology of the brain. Such patients are often misdiagnosed with PD.
ET is a hereditary disease (although sporadic cases are not uncommon) that is characterized by bilateral, symmetrical hand tremors. Unlike parkinsonian tremor, hand tremors in ET are absent at rest and occur during muscle activity. Usually it is most pronounced when the patient stretches his arms in front of him, and can also be present when making voluntary movements, often making everyday activities (for example, eating) difficult. Typically, with ET, there is a “no-no” or “yes-yes” head tremor and voice tremor. The latter may be the first manifestations of ET, and may also occur in isolation in the absence of limb trembling. In contrast to ET, head and voice tremors are generally not found in PD. In contrast, vertical tremor of the mandible at rest and trembling of the legs often occur in PD and rarely in ET. A characteristic sign of ET, which has diagnostic significance, is the disappearance of tremor under the influence of alcohol.
β-blockers also have a good effect on ET. In ET, the “cogwheel” phenomenon may occur, but unlike PD, true akinesia and muscle rigidity are never noted. ET most often affects older people, although it can also occur in very young people.
In the past, vascular damage to the brain was considered the most common cause of P and was even considered a cause of PD. It has now become clear that PD is a primary degenerative disease of the central nervous system and vascular damage does not play an etiological role in its development. Cerebrovascular diseases very rarely cause true P, which was the reason to introduce the concept of “atherosclerotic pseudoparkinsonism” or “vascular pseudoparkinsonism”. Designations such as “lower body parkinsonism” and “parkinsonian ataxia” are also used. Patients suffering from these disorders are typically elderly and often have a history of hypertension. The most characteristic feature of vascular pseudoparkinsonism is a pronounced gait disturbance:
patients take short steps, often spreading their legs wide apart (which is not typical for true P).
Initiation of walking and turning are often difficult, and imbalance and falls also occur. Along with this, any manifestations of P are completely absent in the upper half of the body, arms and face: the patients’ facial expressions are lively, the voice is normally modulated, hand movements are free, and acheirokinesis is absent. Tremor is also not part of the clinical picture. Vascular pseudoparkinsonism can be combined with a decrease in higher mental functions and pyramidal signs (although this is not a mandatory phenomenon). All patients with a similar clinical picture require brain imaging - magnetic resonance imaging
(MRI) or
computed tomography
(CT).
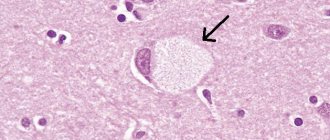
Lewy bodies in Parkinson's disease. In the cytoplasm of the neuron, an eosinophilic nucleus is identified, surrounded by an unstained zone.
The latter, as a rule, reveal multiple small foci of vascular origin (lacunae) in the area of the basal ganglia and/or changes in the white matter of the cerebral hemispheres (leukoaraiosis).
It should be borne in mind that a similar clinical picture can sometimes occur with normal pressure hydrocephalus and less often with tumor lesions of the brain. With normal pressure hydrocephalus, along with walking impairment of the type described above, dementia and dysfunction of the pelvic organs occur (this triad of symptoms is called the Hakim-Adams triad
). MRI of the head typically reveals severely dilated lateral ventricles.
Parkinson's disease
The most common cause of true P (more than 80%), as mentioned above, is PD. This is a degenerative disease in which the main pathological process develops in the nigro-striatal system. In particular, the neurons of the substantia nigra, which produce dopamine, undergo progressive degeneration. As a result, dopamine deficiency develops, which causes clinical symptoms. PD usually affects older people. All the above-described symptoms of P in PD develop fully. PD typically has an asymmetric onset.
At the onset of the disease, symptoms usually affect one side of the body, usually occurring first in the arm (usually the right), then in the leg on the same side.
Often the first manifestation of parkinsonism in the hand is micrographia and acheirokinesis. The hemiparkinsonism stage usually lasts several months, rarely several years. Symptoms then involve the contralateral limbs, as well as the muscles of the trunk and neck. Symptoms of P may appear gradually (for example, akinesia and rigidity in the hand initially appear, then trembling occurs). The opposite situation may also be true. It should be remembered that tremor is absent in 30% of patients with PD at the onset of the disease, and in some cases throughout the entire disease. In this regard, different forms of PD are distinguished
- akinetic-rigid, rigid-tremor and trembling.
At the stage of hemiparkinsonism, the patient’s arm is slightly bent at the elbow joint, does not swing while walking and is pressed against the body. At more advanced stages, when the same changes occur on the other side, and rigidity and akinesia involve the muscles of the trunk and neck, a specific parkinsonian “supplicant pose”
.
Balance problems and falls occur in advanced stages of PD, usually several years after the onset of the disease. PD does not include cerebellar or pyramidal disorders, although there is a tendency for deep reflexes to be increased on the more affected side. Sometimes an extensor position of the foot and toes may be observed, reminiscent of Babinski's symptom, the so-called striatal foot. Symptoms of PD are usually significantly reduced by levodopa
. If parkinsonian symptoms do not respond or respond poorly to sufficient doses of these drugs, the diagnosis of PD should be questioned. With long-term treatment with drugs containing levodopa, almost all patients experience side effects in the form of motor fluctuations and dyskinesias. The latter are expressed as follows: before the next dose of the drug, there is a pronounced increase in P symptoms, often to the point of complete immobility of the patient, and during the period when the concentration of the drug in the blood reaches its maximum values, akinesia and rigidity are completely absent (the “on and off” phenomenon) and are even observed excessive involuntary movements (dyskinesia). The latter are sometimes so pronounced that at first glance at the patient one gets the impression that he has chorea.
The diagnosis of PD is a clinical diagnosis.
Paraclinical research methods usually reveal nonspecific changes. Often, patients with classic PD have vascular changes in the brain. This, however, is in no way evidence of the vascular origin of the disease. It’s just that in most older people, including practically healthy ones, such changes can occur. However, in some cases, atherosclerotic pseudoparkinsonism is superimposed on PD, which may cause a gait unusual for PD and early postural disturbances.
Drug-induced parkinsonism
Drug-induced P may be caused by drugs that act on presynaptic dopamine neurons of the substantia nigra, depleting their dopamine stores (for example, reserpine) or, most often, by antipsychotics that block postsynaptic dopamine receptors, such as phenothiazine derivatives (chlorpromazine), butyrophenones (haloperidol). ), thioxanthines (flupenthixol) and benzamides (sulpiride). These drugs are often used for mental illness. P can also be caused by prochlorperazine (used for vomiting, dizziness and instability), metoclopramide (used for diseases of the gastrointestinal tract, to relieve nausea and vomiting). P may also be due to cinnarizine, which is an atypical calcium channel blocker (used for vestibular disorders). A combination of antipsychotics and antidepressants can also cause P.
In psychiatric hospitals, medicinal P is common. Hypomimia and acheirokinesis are so common among this group of patients that psychiatrists often do not even pay attention to them. Tremor is less common but may have the appearance of classic parkinsonian tremor. Moreover, drug-induced P can be asymmetrical, like PD, and is often completely indistinguishable from PD. One of the signs by which drug P should be suspected is the presence, along with akinetic-rigid syndrome, of violent movements in the form of, for example, oromandibular dyskinesia (involuntary chewing and/or sucking movements) or dystonic phenomena (spasmodic torticollis, oculogyric crises), stereotypy, akathisia (restlessness). Often, severe drug use is accompanied by severe dysarthria and dysphagia. Individual sensitivity to dopamine-blocking drugs is very diverse. Some patients tolerate long-term treatment with large doses of these substances without any problems, while others develop side effects even at small doses. More often
However,
signs of drug-induced P occur when taking large doses of antipsychotics
. Drug-induced P usually develops gradually over days or weeks. In most patients, the first signs appear 3 weeks after the start of treatment. The most common initial signs are hypomimia and insufficient swinging of the arms while walking.
The course of drug P may be different. In most cases, it gradually goes away over several weeks and sometimes days after stopping the drug that caused it. However, it is not uncommon for P to last for months, sometimes almost a year. This situation occurs when using antipsychotic drugs that can be deposited. In rare cases, drug-induced P does not go away and continues to progress despite stopping the causative agent. Such cases are more common among older people. It is believed that in such cases it is not the drug P itself that progresses, but PD begins to develop.
Multiple system atrophy
Multiple system atrophy (MSA) is a sporadic disease that occurs in adults, in which, unlike PD, not only the nigrostriatal system is subject to degeneration, but also many other formations of the central nervous system, including the cerebellum and its connections, pyramidal tracts and formations of the autonomic nervous system (hence the name of the disease). Accordingly, MSA is clinically characterized by a combination of P, cerebellar disorders, pyramidal disorders and progressive autonomic failure
(PVN). P in MSA is caused not only by damage to the cells of the substantia nigra, which causes dopamine deficiency, but also by the degeneration of those postsynaptic receptors with which dopamine must interact.
MSA most often debuts after 50 years of age and, unlike PD, significantly shortens the life expectancy of patients, causing death on average within 9 years from the onset of the first symptoms. P occurs in 90% of patients with MSA, and the dominant clinical sign is in 80%. Cerebellar and pyramidal disorders occur in 50% of patients. Almost all patients show signs of PVN. Autonomic disorders also occur in PD, so one of the key points in the differential diagnosis of MSA and PD is to determine when the signs of PVN began. In MSA, they often occur even before the onset of motor manifestations, often ahead of the latter by several years, while in PD they occur rarely and, as a rule, several years after the onset of the disease. In men, impotence is often the first symptom. A common occurrence among both men and women is urinary incontinence. Often, because of this, patients, before contacting a neurologist, are seen by a urologist and even undergo unnecessary surgical intervention. For comparison: in PD there is only an increase in urination and sometimes an imperative urge to empty the bladder as a result of hyperreflexia of the detrusor muscle. Most people with MSA also experience orthostatic hypotension (OH), which can cause lipothymic conditions and syncope. In patients with PD, OH also occurs, but less frequently and, as a rule, at advanced stages of the disease. Chilliness of the extremities is characteristic - the so-called symptom of cold hands. Approximately 13% of patients with MSA have severe respiratory stridor due to vocal cord abductor paralysis. Stridor initially occurs only during sleep and causes very loud snoring, and as it progresses, it also appears during wakefulness. Vocal cord abductor palsy is so pathognomonic of MSA that, according to some researchers, MSA should be suspected when P is combined with recent severe nocturnal snoring. Syndrome P itself in patients with MSA develops differently than in PD. In particular, MSA P usually begins with a general slowness of movement that affects the arms and legs on both the left and right sides. Although symptoms can also be asymmetrical, the stage of hemiparkinsonism, unlike PD, is usually absent. P phenomena in MSA can begin with dysarthria - the patient’s articulation becomes slower, words are less legible and pronounced less clearly (slurred speech). PD almost never begins with dysarthria. Often, patients with MSA have severe dysphonia - the voice is very muffled and intermittent, often combined with dysphagia, which causes frequent choking while eating. Resting tremor can also occur in MSA, but the classic version of parkinsonian tremor described above, the “coin counting” type, is observed in only 10% of cases. On the contrary, postural trembling and tremor during movement often occur. Irregular myoclonic twitching of the fingers, characteristic of MSA, is often mistakenly regarded as tremor. This disease is also characterized by stimulus-sensitive myoclonus. A number of patients develop so-called disproportionate antecollis
– the head is significantly tilted forward, despite the fact that in general the “bent posture” is not very pronounced. This sign, however, is not considered specific to MSA. It should be remembered that dementia is not part of the clinical picture of MSA. In general, MSA is more malignant than PD and significantly disables patients, often within the first year of the disease.
The clinical picture of MSA may be dominated by one or another symptom. Those cases in which P comes to the fore are referred to as nigrostriatal degeneration
(SND);
if the leading clinical picture is cerebellar syndrome, this condition is called olivo-ponto-cerebellar atrophy
(OPCA);
cases where the core of the clinical picture is PVN are designated by the eponymous name – Shy-Drager syndrome
(SDS).
Despite all the differences, there are often situations when MSA cannot be distinguished from PD. In such cases, one should mainly focus on the effect of levodopa drugs. In PD, these drugs have a dramatic positive effect, while in MSA this effect is less pronounced, short-lived, and often completely absent. This is due to damage to the postsynaptic receptors with which levodopa must interact.
Signs that indicate MSA in patients with P
, the following: rapid progression of symptoms; early onset balance disorders and falls; lack of improvement when treated with levodopa drugs or insufficient effect of these drugs; PVN; pyramidal and/or cerebellar signs; coldness, chilliness of the extremities; contractures; disproportionate antecollis; severe dysphonia, dysphagia or dysarthria; respiratory stridor; irregular tremors or myoclonus.
, special tests for identifying PVN, electromyography of the external ureteral sphincter or anal sphincter can help in diagnosing MSA
which reveals denervation changes in them due to damage to the sacral nucleus of Onuf, from where these muscles are innervated. MRI of the brain often reveals cerebellar atrophy, and with the high resolution of the method (with a magnetic field power of 1.5 Tesla), a hyperintense slit-shaped signal along the outer edge of the putamen is detected in a T2-weighted image. However, none of the listed changes, taken separately, are specific only to MSA, so they should be assessed only in conjunction with the clinical picture. In general, the diagnosis of MSA, like PD, is primarily a clinical diagnosis. During the patient’s lifetime, only “possible” or “probable” MSA is diagnosed on the basis of special diagnostic criteria, and reliable diagnosis is only possible during pathological examination if specific histological markers of the disease are detected - glial cytoplasmic inclusions.
Progressive supranuclear palsy
Progressive supranuclear palsy (PSP), or Steele-Richardson-Olszewski syndrome
, is another variant of degenerations that affects multiple parts of the nervous system, in which the diagnosis of PD is often mistakenly made.
PSP begins later than PD and MSA, most often after 70 years. The average life expectancy after the first symptoms appear is 6–7 years. In this disease, P is characterized by symmetry, akinesia and rigidity in the limbs are not so pronounced, sometimes practically absent, and mainly the axial muscles suffer, i.e. muscles of the trunk and neck. Often, patients with PSP have a slightly thrown back head, although the presence of this sign is not necessary; the head can also be tilted forward or be in a normal position. The first symptom of this disease in most cases is poor balance and falls.
Often, PSP debuts with dysarthria, along with which involuntary deep breaths, similar to a groan, may occur.
The clinical marker of the disease is supranuclear ophthalmoplegia
(NO), or supranuclear gaze paresis, due to damage to specific structures of the midbrain.
OI can be diagnosed when the patient is unable to voluntarily change the direction of gaze, while synkinetic and reflex eye movements are preserved. For example, with PSP, the patient cannot voluntarily move the eyeballs up and/or down, but the Bell
phenomenon - the abduction of the eyeballs upward when closing the eyes;
oculocephalic reflex
(when the gaze is fixed on one point while turning or tilting the head in any direction, the eyeballs are retracted in a friendly manner in the opposite direction).
BUT rarely develops at the onset of PSP; it usually joins other symptoms after an average of 2–4 years. At the beginning of the disease, there may be a pronounced slowing of eye movements, a kind of frozen gaze, burning of the eyes and a feeling of “sand in the eyes” due to the very rare blinking characteristic of PSP. Involuntary forced squinting of the eyes (blepharospasm) is often observed, especially when exposed to bright light or other stimuli. Because of this, some patients are forced to constantly wear sunglasses. so-called apraxia of eyelid opening
, which manifests itself in the fact that it is difficult for the patient to open closed (not necessarily closed) eyes. Due to impaired eye movement, patients may find it difficult to eat because they cannot fully see the plate in front of them. They often drop food on their chest (“dirty tie symptom”). It is difficult for them to go down the stairs; when sitting on a chair, they sit past the seat. All this, combined with a frozen gaze and rare blinking, often gives the erroneous impression that the patient is inadequate, behaves incorrectly, and is inaccessible to contact. Because of this, patients with PSP may sometimes end up seeing a psychiatrist. PSP is also characterized by pronounced pseudobulbar disorders in the form of revitalization of facial axial reflexes; patients often experience a pronounced deepening of the folds on the face (nasolabial, frontal). A pronounced slowdown in mental processes is also characteristic, which is why patients often think for a long time before answering even basic questions.
Thus, although PSP is superficially similar to PD due to the presence of axial muscle rigidity, severe hypomimia and impaired gait, upon careful examination it is revealed that in the distal parts of the limbs PSP is practically absent or very mildly expressed. Classic resting tremor almost never occurs. Levodopa drugs are usually not effective. Autonomic failure is not typical for PSP. MRI of the brain may reveal midbrain atrophy, but this is again a nonspecific finding and the diagnosis of PSP is mainly based on clinical manifestations. Just as in the case of MSA, a diagnosis of “possible” or “probable” PSP is made clinically using specially developed criteria, and a reliable diagnosis is made during pathological examination if specific markers (neurofibrillary tangles) are detected.
Diffuse Lewy body disease
Recently, a new nosological form has begun to be identified that occurs with syndrome P - diffuse Lewy body disease (DLBD). Lewy bodies are intracellular eosinophilic cytoplasmic inclusions that are found in the cells of the substantia nigra in PD and are considered a marker of this disease. In BDTL, they are found not only in the substantia nigra, but in large numbers and widely disseminated throughout the brain. The diagnosis of BDTL is a pathomorphological diagnosis.
Clinically, this disease is characterized by P, which is usually well treated with levodopa, along with dementia with severe visual hallucinations.
Fluctuation in the severity of disorders of higher mental functions
is also typical - mainly due to changes in the ability to concentrate.
Hepatolenticular degeneration (Wilson-Konovalov disease) and other diseases
There are several rarer causes of true or pseudoparkinsonism. One of them, which should always be remembered in the presence of P in people under 45 years of age (including children), is hepatolenticular degeneration, or Wilson-Konovalov disease
.
This is a hereditary disease in which there is a disturbance in the metabolism of copper in the body due to a deficiency of the enzyme ceruloplasmin. As a result, excess copper is deposited in the liver, basal ganglia and around the iris. Wilson-Konovalov disease should be suspected not only if P is present in young people, but also if they have other signs of damage to the extrapyramidal system (for example, dystonia) or mental disorders. Diagnosis is based on detection of copper deposits around the iris using a slit lamp - Kayser-Fleischer ring
.
The latter occurs in 98% of patients at the stage of neurological manifestations. The study of copper excretion in urine and the concentration of ceruloplasmin in the blood
is also of diagnostic importance . The latter, however, in the absence of the Kayser-Fleischer ring and normal copper excretion, has no diagnostic significance. If the situation remains unclear, a liver biopsy or genetic testing is performed. Wilson-Konovalov disease is quite successfully treated with D-penicillamine and zinc supplements in combination with diet.
Toxic P
may occur with manganese intoxication. P caused by the toxin MPTP (1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine) is often encountered, as it selectively affects the substantia nigra, causing the death of dopamine neurons. A toxin similar to this substance is produced during the homemade preparation of certain narcotic substances, so P is often observed among young people suffering from drug addiction. Along with P, pyramidal and pseudobulbar symptoms may be observed.
P occurs in boxer encephalopathy
. It is believed that in this case, the etiological role is played by regularly received multiple craniocerebral injuries. It should be remembered that boxer encephalopathy is practically the only situation where traumatic brain injury plays a role in the development of P. In general, in people who are not involved in boxing, the presence of a history of traumatic brain injury (one or more) is not given etiological significance.
P may sometimes occur with Huntington's chorea
, which usually occurs with pronounced hyperkinesis. This hereditary disease usually debuts in the fourth decade of life, but there are cases of earlier onset. Usually, the so-called juvenile form of Huntington's chorea (Westphal form) occurs with akinetic-rigid syndrome. There are also familial cases of P, including juvenile cases. Therefore, in all patients with P syndrome, collecting a hereditary history is of great importance.
Akinesia and rigidity may sometimes be observed in Alzheimer's disease, Pick's disease,
in which the core of the clinical picture is progressive dementia.
In Alzheimer's disease, memory is primarily affected, and in Pick's disease, personality disorder comes to the fore.
In these diseases, P is often manifested by moderate akinesia, rigidity is less pronounced, and parkinsonian tremor is almost never observed.
Corticobasal degeneration
(CBD) is one of the very rare causes of P. With CBD, akinesia and rigidity are strictly asymmetrical, usually present in one arm and accompanied by myoclonus in it, dystonic phenomena and apraxia.
A characteristic sign of the disease is “alien hand syndrome”
. The latter is manifested by the fact that the patient’s hand, regardless of his desires and intentions, makes various movements, sometimes even quite complex motor acts.
Postencephalitic P
nowadays it practically never occurs. Many cases have been observed since the encephalitis lethargica pandemic at the beginning of the 20th century. Since then, only a few cases have been reported in the literature.
A special endemic form of P is observed on the island of Guam
and in some other places in the eastern Pacific. In these places, there are many cases of P in combination with amyotrophies and dementia (Guam parkinsonism-dementia complex). Pathomorphologically, the brains of these patients look like the brains of patients with PSP. The histological markers are also the same.
Treatment
People with MSA are treated with symptomatic medications. They help eliminate the severity of parkinsonism, cerebellar ataxia and other signs of brain degeneration.
Symptomatic treatment of multiple system atrophy includes:
- levodopa medications;
- vasoactive agents;
- neurometabolic drugs;
- massage, water procedures, exercise therapy, other methods of physiotherapy;
- diet in compliance with salt intake;
- non-drug ways to eliminate orthostatic hypotension.
Parkinsonism in the initial stages of MSA progression is treated with combination drugs of levodopa with benserazide, carbidopa. It is replaced if it is poorly tolerated or ineffective with alternative therapy drugs. For atrophy of all types, dopamine receptor agonists and medications with the substance amantadine are used.
Manifestations of cerebellar ataxia are eliminated using physiotherapy methods. To alleviate the condition of MSA, medications containing clonazepam, gabapentin, buspirone, propranolol and other substances are prescribed.
For pelvic disorders against the background of atrophy, medications are used:
- Sildenafil (for erectile dysfunction);
- Macrogol, laxatives (in case of constipation);
- Alpha1-adrenergic receptor antagonist + cholinergic drug (urinary retention);
- Anticholinergic (involuntary or painful urination).
In case of bladder dysfunction, in addition to medications, periodic or continuous catheterization of the organ is indicated. Alternative medications include botulinum toxin injections. These injections are also prescribed for the treatment of respiratory disorders, captocormia, drooling, and dystonia due to MSA.
For orthostatic hypotension, it is recommended to sleep on beds with an elevated headboard and drink plenty of water rich in minerals. You must wear compression stockings. You can’t overeat or get up suddenly after waking up. It is recommended to learn isometric maneuvers. Mineralocorticoids and hypertensive drugs with midodrine are prescribed.
In case of breathing problems, ventilation or tracheostomy is indicated. Camptocormia is treated with physical therapy. Medicines with zopiclone or clonazepam eliminate sleep disorders due to MSA. For depression, a selective serotonin reuptake inhibitor (a group of antidepressants) is prescribed.
Reference! Clinicians continue to seek new treatments for multifocal atrophy. Etiological therapy cannot be used until the causes and mechanism of development of MSA are elucidated.
Complications
The consequences of MSA progression include urogenital infections, cystitis, urethritis, and kidney inflammation. Bacteria can enter the blood and cause sepsis. Against the background of respiratory disorders, pneumonia and sleep apnea often develop.
With multiple system atrophy, damage to the medulla oblongata is possible. This is complicated by a disorder of swallowing function and causes death due to paralysis of the respiratory center or asphyxia. As a result of degenerative processes, the functioning of the heart, vascular system, and cerebral circulation is also disrupted.
Prognosis and life expectancy
MSA is an incurable pathology with irreversible changes in the brain. Therefore, the prognosis for recovery is negative. Treatment alleviates the manifestations of multiple system atrophy, but is not able to stop or slow down the degeneration of neuroglia. The quality of life is rapidly deteriorating, a person cannot take care of himself.
Death occurs on average 7 years after the first signs of the disease appear. With slow progression, life expectancy doubles. Death is caused by complications of MSA: infections, cardiac or respiratory failure, cerebrovascular accident.
Forecast
Treatment is exclusively symptomatic.
It is based on levodopa and other antiparkinsonian drugs. Multiple system atrophy is a currently incurable disease with a steadily progressive course. Patients' quality of life quickly deteriorates, and 5–7 years after the onset they may no longer be able to move independently and lead a semi-bed lifestyle.
As the disease progresses, complications arise. Most often noted:
- recurrent urogenital infection;
- bronchopneumonia against the background of increasing respiratory failure;
- bulbar syndrome with swallowing disorders;
- apnea (stopping breathing) during sleep;
- gross disturbances in the functioning of the cardiovascular system;
- acute cerebrovascular accidents due to episodes of hypotension or due to nocturnal hypertension.
The life expectancy of patients with multiple system atrophy after the development of the main symptoms is short, although cases of relative stabilization of the condition for several years have been described. Early appearance and rapid progression of autonomic failure worsens the prognosis. Mortality is usually associated with acute vascular disorders, sepsis, pneumonia, and apnea.
Prevention methods
Since the causes and pathogenesis of MSA are not fully understood, specific prevention has not been developed. Doctors recommend using respirators and other personal protective equipment when working with neurotoxic substances.
To prevent genetic predisposition, a pregnant woman is strictly prohibited from smoking, using drugs, alcohol, or taking medications with teratogenic and embryotoxic properties. Medicines are prescribed only by a doctor for strict medical reasons.
What do you need to remember?
- With multiple system atrophy, irreversible degeneration of central nervous system tissue occurs in the brain.
- The causes of occurrence and the mechanism of development continue to be studied.
- In the ICD-10 reference book, two types of the disease were left: parkinsonian and cerebellar.
- In the MSA clinical picture, 2–3 syndromes are present simultaneously: autonomic failure in combination with cerebellar dysfunction and/or parkinsonism.
- During the diagnosis, the patient is tested for various types of central nervous system disorders and undergoes brain tomography.
- Treatment of multifocal brain atrophy involves the use of symptomatic therapy.
- Sepsis, bulbar palsy and other complications of MSA lead to death.
- There is no prognosis for recovery; the patient lives a maximum of 15 years.
- Prevention of MSA has not been developed because the causes of the disease are unclear.
Literature
- Shindryaeva N.H., Belova A.N., Levin O.S. Multiple system atrophy is common in Nizhny Novgorod. Collection of materials of the scientific and practical conference “Autonomic disorders in the clinic of nervous and internal diseases -2009” 2009
- Shindryaeva N.H., Levin O.S. Autonomic manifestations in patients with multiple system atrophy. Collection of materials of the III scientific and practical conference “Autonomic disorders in the clinic of nervous and internal diseases” 2010.
- Damulin I.V., Yakhno HH, Goncharov OA Comparative assessment of disorders of higher brain functions in various types of cerebral atrophy.// Journal. neurology and psychiatry S.S. Korsakov -1990.
- Ponomarev, V.V. Rare neurological syndromes and diseases. -SPb. Folio.
- Brown RG, Lacomblez L, Landwehrmeyer BG, Bak T, Uttner I, Dubois B, Agid Y, Ludolph A, Bensimon G, Payan C, Leigh NP; for the NNIPPS Study Group (August 2010). “Cognitive impairment in patients with multiple system atrophy and progressive supranuclear palsy.” Brain. 133 (Pt 8): 2382–93.
- Swan L, Dupont J (May 1999). “Multiple system atrophy.” Phys Ther. 79(5): 488–94. PMID 10331752.
- American Psychiatric Association. Diagnostic and statistical manual of mental disorders, 4th edition, (DSM IV). // Washington, DC: American Psychiatric Association, 1994.
- The Consensus Committee of the American Autonomic Society and the American Academy of Neurology (1996). “Consensus statement on the definition of orthostatic hypotension, pure autonomic failure, and multiple system atrophy.” Neurology. 46(5): 1470. PMID8628505.