О заболевании
Множественной системной (мультифокальной, мультисистемной) атрофией называют обширную дегенерацию глиальных клеток во всех структурах головного мозга (ГМ). Сокращенно болезнь записывают МСА или MSA. Мультисистемная атрофия быстро прогрессирующее тяжелое заболевание, при котором в патологический процесс часто вовлекается спинной мозг, приводя к уменьшению тканей центральной нервной системы инвалидности.
Дегенерация провоцирует появление:
- слабоумия;
- дрожательного паралича — синдром паркинсонизма;
- дисфункциивегетативной нервной системы;
- атаксии мозжечка — расстройство двигательной и речевой функции;
- недостаточности пирамид — нарушается работа ЦНС.
Справка! МСА является редкой неизлечимой патологией, встречается преимущественно у мужчин. Заболеваемость составляет менее 6 случаев на 100000 населения.
Во время развития болезни поражаются подкорковые ядра (базальные ганглии) внутри полушарий ГМ.Они расположены между промежуточным мозгом и лобными долями. Состоят из полосатого тела, серого и белого вещества, нигральной (черной) субстанции, субталамического ядра. На гистологическом исследовании в олигодендроглиоцитах обнаруживаются накопления альфа-синуклеина во включениях цитоплазмы. Это несвойственно составу клеток нейроглии.
В результате дегенерации подкорковых ядер нарушается:
- координация движений;
- регуляция мышечного тонуса;
- чувствительность восприятия зрительных, слуховых и прочих раздражений;
- регулирование трофики, обмена, дыхания, мочеиспускания, иных вегетативных функций;
- выработка рефлексов, память, другая регуляция высшей нервной деятельности.
МСА чаще выявляют у людей старше 50 лет, которые работали с вредными и токсическими веществами, пестицидами, формальдегидом, растворителями. В группе риска состоят пациенты с болезнью Паркинсона, Альцгеймера. Опасность заключается в том, что патология вызывает необратимый процесс в мозге с гибелью нервных клеток. С момента появления симптомов длительность жизни не превышает 15 лет. Смерть чаще наступает вследствие нарушения дыхания или сепсиса.
Рисунок 1. Отображение мозга при томографии
До 2021 года в МКБ―10 мультисистемная атрофия была причислена к коду G90.3 под названием «полисистемная дегенерация». Сейчас в справочнике остался паркинсонический тип МСА под шифром G23.2 «MSA-P» и мозжечковый – G23.3 «MSA-C». В МКБ-10-КМ патология о под номером G90.3.
Магнитно-резонансная томография (МРТ) в Санкт-Петербурге
МРТ головного мозга. Демонстрация атрофии и нормы при цветовой обработке.
Под термином “нейродегенеративные заболевания” (НДЗ) определяется большая группа заболеваний преимущественно позднего возраста, для которых характерна медленно прогрессирующая гибель определенных групп нервных клеток и одновременно – постепенно нарастающая атрофия соответствующих отделов головного и/или спинного мозга. В основе развития этих заболеваний лежит нарушение метаболизма и изменение конформации клеточных белков с их последующим накоплением и агрегацией в определенных группах нейронов. При НДЗ страдают преимущественно нейроны и глиальные клетки базальных ганглиев и стволовых структур, вырабатывающие ацетилхолин, дофамин, серотонин.
Классификация делит НДЗ на 2 большие группы – спорадические и ирритативные.
- Спорадические НДЗ:
- Прогрессирующий надъядерный паралич (болезнь Стила —Ричардсона — Ольшевского).
- Мультисистемная атрофия.
- Деменция с тельцами Леви.
- Паркинсоническая деменция (синдром Гуам).
- Кортикобазальная дегенерация.
- Болезнь Альцгеймера.
- Ирритативные НДЗ:
- Болезнь Гентингтона.
- Болезнь Галлервордена—Шпатца.
- Болезнь Вильсона—Коновалова.
- Болезнь Фара.
- Болезнь Бессена — Корнцвейга.
Болезнь Альцгеймера – прогрессирующее нейродегенеративное заболевание, характеризующееся постепенным развитием деменции. Происхождение заболевания точно неизвестна. Биохимические изменения состоят в снижении активности холин-ацетил-трансферазы коры головного мозга и гиппокампов. Патологические проявления заключаются в образовании специфических амилоидных бляшек, нейрофибриллярных тяжей и реактивном глиозе. Развивается атрофия, захватывающая преимущественно кору вокруг Сильвиевых щелей и гиппокампы, с вторичным расширением желудочков, особенно височных рогов
Заболевание впервые описано Alois Alzheimer в 1907 году. Процесс напоминает естественное старение, но резко ускоренное. Начинается с нарушений памяти, затем потерянность, невозможность повседневного самообслуживания, повторяющиеся вопросы. Позже присоединяются глубокие нарушения психики, речи, потеря веса, судороги.
Частота составляет 0,51% для лиц в возрасте 70-74 лет с возрастным прогрессирующим увеличением частоты. Клинические проявления состоят в нарушении памяти, депрессии, поведенческих нарушениях и галлюцинациях. На поздних стадиях к психическим расстройствам добавляется экстрапирамидная симптоматика. Заболевание занимает 4 место по смертности. Диагноз ставится на основании клинического и нейрофизиологического обследования, а также нейровизуализации. Типичные проявления на КТ состоят в диффузной атрофии (особенно,височных долей), вторичном расширении борозд и желудочков. Чувствительность (без измерения объемов) в сравнении с нормальной возрастной группой около 80%, специфичность около 70%. Измерение объемов гиппокампов при выполнении МРТ с тонкими срезами повышает точность до 85%.
МРТ головного мозга. Т2-взвешенная сагиттальная МРТ. Болезнь Пика. Цветовая обработка изображения.
МРТ головного мозга служит метода выбора оценки структурных изменений. Атрофические изменения выражены во всём медиобазальном отделе височной доли. Чувствительность и специфичность МРТ при начальной деменции около 80%. Измерение объемов гиппокампов и амигдалы повышает точность до до 85%.
МРТ головного мозга. Т1-взвешенная корональная МРТ. Диффузная атрофия при болезни Альцгеймера.
Дифференциальную диагностику при МРТ головного мозга надо проводить с болезнью Паркинсона, мультиинфарктной деменцией и лобнотеменной деменцией (болезнь Пика).
В этих же зонах отмечается гипоперфузия и снижение активации при фМРТ. Кроме МРТ при исследовании болезни Альцгеймера важное значение имеет ПЭТ с [18F] флюоро-2-деоксиглюказой (FDG). Гипометаболизм хорошо коррелирует с тяжестью заболевания и предсказывает его развитие.
Синдромы Паркинсона включают группу заболеваний, близких по клинике к болезни Паркинсона. К синдромам Паркинсона относится быстро прогрессирующая деменция с тельцами Леви . При МРТ головного мозга низкий сигнал наблюдается не только от компактной части черного вещества, но и от скорлупы, которая становится даже темнее бледного шара. При оливопонтоцеребеллярной атрофии на сагиттальных МРТ головного мозга видно уменьшение объема моста и мозжечка. При прогрессирующем надъядерном параличе обнаруживается атрофия пластины четверохолмия. Описаны характерные симптомы при МРТ – «пингвина», «Микки Мауса» и другие, смысл которых заключается в описании признаков атрофии.
При деменции, связанной с болезнью Паркинсона при МРТ головного мозга отмечается снижение толщины коры в проекции парагиппокампальной части левой средней затылочно-височной извилины и уменьшение объема левого нижнего продольного пучка. Уменьшение толщины коры левой парагиппокампальной зоны связано с высоким риском депрессии. Отмечено, что дневная сонливость коррелирует с уменьшением толщины коры фузиформной зоны, определяемое при МРТ.
Для прослеживания динамики и прогнозирования также прибегают к различным измерениям при МРТ головного мозга:
- коэффициент средний мозг-мост в норме 0,24, а при прогрессирующем надъядерном параличе становится меньше 0,12.
- Индекс паркинсонизма – отношение ширины верхней ножки мозжечка в корональной плоскости к площади среднего мозга в средней сагиттальной плоскости умноженной на отношение ширины средней ножки мозжечка к ширине верхней ножки мозжечка – больше 13,55 свидетельствует в пользу паркинсонических синдромов. При МРТ выявляется атрофия хвостатых ядер с вторичным расширением передних рогов; атрофия скорлупы и коры лобных долей. Отношение ширины передних рогов к расстоянию между хвостатыми ядрами (по их краям), измеряемое в поперечной плоскости уменьшается с 2,2-2,6 до значений близких к 1,0. Другой коэффициент – расстояние между хвостатыми ядрами (между их головками к ширине черепа по внутренним пластинкам) – увеличивается (в норме 0,09-0,12).При МРТ головного мозга выявляется диффузная атрофия мозга, расширение периваскулярных пространств Вирхова- Робена и лейкоараиоз. Последний является следствием стеноза и окклюзии глубоких вен мозга. На Т2-зависимых МРТ изображениях лейкоараиоз выглядит как небольшие очаги гиперинтенсивности. В целом эти признаки неспецифические и отражают старение мозга. При МРТ головного мозга на томограммах обоих типов взвешенности обнаруживается повышенный сигнал от моста и покрышки мозжечка. Типично изменение сигнала от периферии моста. В отличии от опухолей при МРТ нет отека и масс-эффекта. Самые ранние проявления обнаруживаются на диффузионное-взвешенных МРТ головного мозга, примерно через 24 часа от начала тетрапареза.
Прогрессирующий надъядерный паралич проявляется в виде нарушения взора вверх, экстрапирамидной симптоматики и умственных нарушениях. Заболевание развивается у лиц около 60 лет. Этиология неизвестна, почти все случаи спорадические. Частота 1-1,5 случаев на 100 тыс. населения. Заболевание характеризуется патологическим скопление в головном мозге тау-протеина. При МРТ головного мозга наблюдается диффузная атрофия, причем на сагиттальных Т1-взвешенных МРТ отмечается характерный симптом “пингвина”. Атрофические изменения моста и среднего мозга приводят к расширению водопровода и III желудочка, контур которых напоминает очертания пингвина.
МРТ головного мозга. Т12-взвешенная сагиттальная МРТ. Прогрессирующий надъядерный паралич. Симптом “пингвина”.
Центральный понтинный миелиноз (осмотическая деменция) представляет собой приобретенное метаболическое расстройство. Чаще встречается у алкоголиков. Гипонатрийемия приводит к демиелинизации, видимой при МРТ. Центральный понтинный миелиноз часто сопровождается экстрацентральным, когда есть поражение выше ствола. Клинические проявления сводятся к заторможенности (вплоть до летаргии), спастическому тетрапарезу и поражению нижних черепных нервов.
МРТ головного мозга. Т2-взвешенная аксиальная МРТ. Центральный понтинный миелиноз,
Болезнь Бинсвангера (субкортикальная атеросклеротическая энцефалопатия, деменция мелких сосудов). Это состояние, связанное с множественными инфарктами мелких ветвей, что при МРТ головного мозга видно как лакунарные ОНМК. Заболевание постепенно прогрессирует. Вариантом болезни Бинсвангера можно считать наследуюмую семейную артериопатическую лейкоэнцефалопатию.
МРТ головного мозга. Т2-взвешенная МРТ типа FLAIR. Болезнь Бинсвангера.
Болезнь Гентингтона относится к наследуемым заболеваниям, проявляется в среднем возрасте и быстро прогрессирует. В клинической картине преобладают хореоатетоз и деменция.
МРТ головного мозга. Т1-взвешенная корональная МРТ. Болезнь Гентингтона.
Болезнь Фара очень редкое наследственное заболевание, проявляющееся в кальцификации базальных ганглиев и зубчатого ядра. На Т2-зависимых МРТ ядра резко гипоинтенсивны, что соответствует кальцинатам, хорошо видимым на КТ. Нередко в области зрительных бугров обнаруживаются мелкие гиперинтенсивные очажки.
МРТ и КТ головного мозга в аксиальной плоскости. Болезнь Фара.
Об МРТ в СПб при нейродегенераторных заболеваниях можно также читать на странице нашего другого сайта. Исследование мы выполняем в закрытом аппарате, но можно и на открытом МРТ. МРТ СПб позволяет выбирать место МРТ, но в этом случае мы рекомендуем Вам обследоваться у нас.
Оставить отзыв.
МРТ в Санкт-Петербурге USA
Причины развития
Ученые продолжают изучать причины, механизм развития и провоцирующие факторы МСА. Однозначного подтверждения нет о наследственной предрасположенности к развитию болезни. У ребенка врожденная атрофия ГМ бывает при злоупотреблении женщины во время беременности лекарствами, спиртным, наркотиками.
В ходе диагностики врачи выявляют возможные причины мультисистемнойатрофии:
- болезнь Паркинсона либо Альцгеймера;
- контакт с нейротоксическими веществами;
- изменчивость гена «α-синуклеин»;
- отравление алкоголем, наркотиками у людей с зависимостью;
- травма головного и/или спинного мозга;
- гипоксия тканей ГМ.
Патогенез плохо поддается изучению, поскольку неизвестны точные причины мультисистемной атрофии. В ходе исследований обнаружено накопление тау-протеинав пораженных олигодендроглиоцитах. Это выявляют в мозжечке, пирамидах, коре ГМ, рогах спинного мозга в области грудного и крестцового отдела хребта. Одновременно повреждаются дофаминовые рецепторы, черная субстанция, скапливаетсяα-синуклеин в нейроглиальных клетках.
Справка! МСА характерно асимметрическое уменьшение белого вещества, нарушение передачи нервных импульсов. Нейроны страдают меньше олигодендроглиоцитов.
Классификация
Медики выделяют три формы МСА в зависимости от того, какой синдром выявляется ведущим. Если это невозможно установить, пациенту диагностируют смешанный тип болезни.
Классификация патологии:
| Ведущий симптом | Тип мультисистемной атрофии | Отличительные черты |
| Паркинсонизм | Стриатонигральный тип МСА | Замедленные движения, лицо маскообразное, застывание в одной позе, симптом «воздушная подушка», тремор конечностей, согнутость суставов, снижение подвижности. Дегенерации больше подверженстриатум, черная субстанция. |
| Вегетативная недостаточность | Синдром Шая-Дрейджера | Дисфункция желез и органов, тазовые нарушения, гипотензия, храп, апноэ. |
| Мозжечковая атаксия | Оливопонтоцеребеллярный тип МСА | Ухудшение равновесия, нарушение мелкой моторики, непроизвольное движение глазных яблок, мышечная слабость. Дегенерации больше подвержен мозжечок, мост, оливы. |
В классификаторах врачи предлагают убрать синдром Шая-Дрейджера, поскольку вегетативная недостаточность сопровождает все формы МСА. В МКБ-10 указан только мозжечковый и паркинсонический тип болезни.
Клинические разновидности
В зависимости от комбинации и преобладания симптомов выделяют несколько клинических типов мультисистемной атрофии:
- Стрионигральная форма с доминированием паркинсонизма. Его проявления маскируют другую, менее выраженную симптоматику. В отличие от болезни Паркинсона, при этом обычно нет периода гемипаркинсонизма с односторонностью тремора и ригидности. Дрожание может комбинироваться с миоклоническими подергиваниями мелких мышц, приобретая аритмичный и неравномерный характер. Характерна малая чувствительность к дофамин-содержащим препаратам, а возникающие поначалу эффекты от лечения быстро угасают.
- Оливопонтоцеребеллярная форма, с преобладанием мозжечковых расстройств.
- Вегетативная форма, или синдром Шая-Дрейджера. Выраженная и мало корректируемая ортостатическая гипотензия становится основным фактором инвалидизации, уже на ранних стадиях болезни приводящим к социально-бытовой дезадаптации. Ее диагностируют, если в вертикальном положении систолическое давление снижается на 20 и более мм рт.ст, а диастолическое – на 10 и более мм рт.ст. (по сравнению с уровнем в положении лежа). Характерны жалобы на утомляемость, несистемное головокружение, предобморочные и синкопальные состояния, акроцианоз. Ортостатическая гипотензия может дополняться ночной артериальной гипертонией, малой адаптивностью пульса к нагрузкам.
Чаще всего встречается комбинация паркинсонизма с вегетативными нарушениями, причем по мере развертывания симптоматики тип болезни может меняться. Добавление мозжечковых расстройств усугубляет состояние, в наибольшей степени сказываясь на возможности самостоятельного безопасного передвижения.
Симптомы
Первый признак МСА – начало прогрессирования болезни в старшем возрасте после 45 лет. Симптомы развивается быстро. У большинства людей сразу проявляется паркинсонизм, двигательные нарушения. В 40% случаев дегенерация стартует с вегетативной дисфункции.
Изменения в начальной стадии пациентом не всегда замечается. Среди первых признаков указаны тазовые нарушения: эректильная дисфункция, трудности с мочеиспусканием или дефекацией, недержание мочи/кала. Каждый пятый заболевший с момента прогрессирования мультисистемной атрофии начинает падать. Причиной считается ортостатическая гипотензия, мышечная слабость, дисфункция мозжечка.
Справка! Прогрессированию МСА характерно присоединение к ведущему синдрому других симптомокомплексов. То есть, у человека одновременно проявляется вегетативная недостаточность в сочетании с признакамипаркинсонизма и мозжечковой атаксии.
Отличия симптомов разных типов МСА
Первичные симптомы зависят от класса мультисистемной атрофии. При стриатонигральном типе МСА сразу заметны признаки болезни Паркинсона. Вначале организм откликается на лечение леводопой, затем эффективность лекарств теряется, усугубляются вегетативные расстройства.
Первичные признаки паркинсонического типа мультисистемной атрофии:
| Симптом | Пояснение |
| Брадикинезия | Все произвольные движения замедляются. Человек медленнее ходит, говорит, пишет, читает вслух. Координация движений и речи сохраняется. Длительный разговор или необходимость движения вызывает быструю утомляемость. |
| Ригидность | Скованность движения, напряжение мышц, отвечающих за сокращение, разгибание. Подбородок почти касается ключичной зоны. В горизонтальном положении на спине голова не лежит на подушке, но это исчезает после засыпания. Конечности полусогнуты в крупных суставах, туловище сгибается вперед, позвоночник сутулый. При пассивном движении конечностью (выполняет доктор) под пальцами врач ощущает вязкое сопротивление мышц. |
| Постуральная неустойчивость | Человек не может сохранить равновесие. Это не связано с ортостатической гипотонией, потемнением в глазах, гипертензией. |
| Тремор | Мышцы туловища, шеи, рук, ног дрожат во время движения или покоя. Тремор исчезает, когда пациент выполняет противоположное действие. То есть, перестает либо начинает двигаться. |
При оливопонтоцеребеллярном типе МСА на первом плане стоят симптомы мозжечковой дисфункции. Пациент начинает семенить (уменьшается длина шага). Отмечается шаткость походки, скованность мышц, ухудшение общей координации движений и мелкой моторики. Тремор усиливается при приближении к цели движения. Затрудняется смена быстро чередующихся действий.
При мозжечковом типе мультисистемной атрофии проявляется дизартрия и глазодвигательная (окуломоторная) дисфункция. Их симптомы:
- Приглушенность голоса;
- Растянутое произношение слов;
- Скандированная речь;
- Нарушение модуляция звука, фонации, дыхания во время произношения;
- Ритмичное непроизвольное движение глазных яблок (нистагм).
Синдром Шая-Дрейджера при МСА проявляется расстройством функций тазовых органов, желез. Бывает обморок, коллапс из-за падения давления. К признакам относят нарушение мочеиспускания, опорожнения кишечника, снижение слюнотечения, слезотечения, потоотделения. Отмечается во время сна движения глаз, разговор, кратковременная остановка дыхания. У мужчин ухудшается эрекция, развивается импотенция.
Справка! Прогрессирование МСА проявляется усугублением симптомов 1―3 типов мультисистемной атрофии. Клиника дополняется слабоумием, параличом или парезом, неадекватным поведением, осложнениями дегенерации.
Международный неврологический журнал 5 (51) 2012
Паркинсонизм — прогрессирующее заболевание нервной системы, характеризующееся замедленностью произвольных движений, ригидностью мышц, их дрожанием (тремором) в состоянии покоя, обеднением мимики, изменением походки (маленькие шажки, отсутствие нормального размахивания рук). Наиболее частой формой паркинсонизма является идиопатический паркинсонизм (или болезнь Паркинсона), главные отличительные проявления которого — ригидность, тремор покоя, нарушение постуральных рефлексов, асимметрия начала, хорошая реакция на препараты леводопы, гибель клеток черной субстанции и наличие телец Леви [1, 2]. Но существует несколько других первичных нейродегенеративных заболеваний с поражением экстрапирамидной системы, при которых синдром паркинсонизма (табл. 1) может быть одним из основных или дополнительных проявлений [5, 7, 8].
На ранних стадиях заболевания существуют трудности дифференциальной диагностики различных форм паркинсонизма. Диагностику разных паркинсонических нарушений существенно улучшает использование специальных критериев, таких как система оценки степени тяжести паркинсонизма Л.С. Петелин с сотр. (1980), шкала Хен — Яра (1967) в модификации Lindval с сотр. (1989), Tetrud, Langstone (1989), унифицированная шкала оценки болезни Паркинсона (Fahn, Elton, 1987), критерии диагностики болезни Паркинсона общества БП Великобритании (Gibb, Lees,1988), критерии диагностики мультисистемной атрофии (Gilman et al., 1998), критерии диагностики прогрессирующего надъядерного паралича (критерии NINDSSPSR Litvan et al., 1996), клиниконейровизуализационные критерии диагностики сосудистого паркинсонизма (Левин О.С., 1997), критерии диагностики эссенциального тремора (Elble, 2000), критерии диагностики синдрома Туретта (The Tourette Syndrome Classification Study Group, 1993) и др. [8–10].
Использование сочетания критериев по разным шкалам позволило нам уже на стадии клиники провести дифференциальную диагностику этиологического фактора синдрома паркинсонизма, наблюдаемого нами у 60летнего больного.
В августе 2011 г. в отделение ангионеврологии и нейрореабилитации ГУ «Институт неотложной и восстановительной хирургии им. В.К. Гусака НАМН Украины» поступил пациент Р., 60 лет, с жалобами на скованность в руках и ногах, слабость в них, шаткость при ходьбе, замедленность походки, неуклюжесть, замедление речи, недержание мочи, потемнение в глазах при перемене положения тела, склонность к падениям, снижение памяти на недавние события.
Болеет около 10 лет, когда стал замечать неустойчивость при ходьбе, слабость в ногах. Значения этому не придал. Заболевание медленно прогрессировало. В течение последних 3 лет отмечает значительное ухудшение состояния: выраженная скованность в ногах, затруднение ходьбы, шаткость при ходьбе. Наблюдался у невролога по м/ж с диагнозом: дисциркуляторная энцефалопатия (атеросклеротическая) 1–2й ст. с паркинсоническим синдромом, акинетикоригидная форма, прогредиентное течение с рефлекторнопирамидной недостаточностью в конечностях. Принимал мирапекс, левоком, наком. Эффекта от терапии практически не отмечал. Наследственный анамнез не отягощен. Направлен в отделение в связи с отсутствием эффекта от лечения и появлением вегетативных нарушений (потливость, низкое АД, слабость, головокружение, недержание мочи, синкопе, т.е. симптоматика пандизавтономии).
Объективно: состояние относительно удовлетворительное. Повышенная сальность кожи лица с наличием гиперемии в области носогубного треугольника, лба и бровей, шелушение кожи. Мраморный рисунок кожи стоп. Периферические л/у не увеличены. В легких дыхание везикулярное, хрипов нет. Глухие тоны сердца. АД 110/70 мм рт.ст. Ритм сердца — 64 в 1 мин. Живот мягкий, чувствительный по ходу толстого кишечника, в области белой линии грыжевое выпячивание при натуживании. Отеков нет. Симптом Пастернацкого отрицательный.
Неврологический статус: гипомимия, брадикинезия, брадилалия. Мышечный тонус повышен по экстапирамидному типу — вязкий, пластический, монотонный, усиливается при каждом повторном пассивном движении (феномен «восковой куклы»), симптом Нойка с 2 сторон. Постуральный тремор конечностей, постуральная неустойчивость. Ахейрокинез. Глазные щели D < S, зрачки равны, реакции на свет живые. Слабость отведения во все стороны, конвергенции. Горизонтальный нистагм при крайних отведениях. Мягкое небо малоподвижно. Небные рефлексы не вызываются. Язык по средней линии. Дисфония. Дизартрия. Положительные рефлексы орального автоматизма. Рефлексы с рук высокие S > D. Коленные повышены D > S, ахилловы повышены D > S. Брюшные рефлексы снижены. Патологические кистевые рефлексы. Глубокая, болевая чувствительность не нарушена. Вибрационная чувствительность на лодыжках D — 12 с, S — 14 с. Координаторные пробы выполняет с дисметрией и интенционным тремором. Асинергия Бабинского, симптом Стюарта — Холмса. Походка шаркающая, с широко расставленными ногами.
При исследовании функции вегетативной нервной системы выявлены:
— сердечнососудистые нарушения в виде наличия ортостатической гипотензии (80/50 мм рт.ст.), артериальной гипотензии после еды (90/60 мм рт.ст.), артериальной гипертензии в положении лежа, снижения вариабельности сердечного ритма;
— желудочнокишечные нарушения в виде наличия жалоб на дискомфорт в эпигастрии после еды, гиперсаливацию, тошноту, запоры и ощущения неполного опорожнения кишечника при акте дефекации;
— мочеполовые нарушения в виде расстройств мочеиспускания по ирритативному типу, нарушение потенции;
— трофические нарушения в виде сухости, истончения кожи и наличия себорейного дерматита;
— нарушение зрачковой иннервации в виде жалоб на нечеткость зрения в темных или ярко освещенных помещениях;
— сетчатое ливедо на коже стоп.
Дополнительные обследования: общеклинические и биохимические анализы крови, мочи и ликвора, электролиты, сахар крови без патологии. Холестерин крови — 4,7 ммоль/л, Среактивный белок — 2 мг/л, ревматоидный фактор — 8 МЕ/мл, АСЛО — 54 Ед/мл, церулоплазмин крови — 0,25 г/л, медь в суточной моче — 28 мкг/сут, адреналин в моче — 14 мкг/сут, норадреналин в моче — 45 мкг/сут, дофамин в моче — 438 мкг/сут. Общий белок — 75 г/л, альбумины — 43 г/л, a1глобулины — 5,8 %, a2глобулины — 8,7 %, bглобулины — 10,1 %, гаммаглобулины — 16,4 %. Ликворное давление составило 140 мм вод.ст.
МРТ головного мозга — высокая интенсивность сигнала в Т2режиме в области моста и средних мозжечковых ножках отражает дегенерацию понтоцеребеллярных волокон, снижение интенсивности сигнала в области скорлупы. Уменьшение объема коры в первичной сенсорномоторной, латеральной премоторной и префронтальной областях, уменьшение объема мозжечка.
МРТ шейного отдела позвоночника и спинного мозга 14.09.11 — остеохондроз шейного отдела позвоночника с протрузиями дисков С4С5, С5С6, С6С7. Деформирующий спондилез, спондилоартроз. Дополнительных образований и очагов патологически измененного МРсигнала в спинном мозге и экстрамедуллярных пространствах не выявлено.
Дуплексное сканирование магистральных артерий головы — просвет ОСА, ВСА чистый, внутрипросветных образований не выявлено, дифференцировка артериальной стенки на слои сохранена, КИМ 1,1 мм, умеренная гемодинимически незначимая Sобразная извитость ВСА с 2 сторон.
ЭКГ — синусовый ритм, вертикальное положение ЭОС, ЧСС 78 в 1 мин.
Уролог — нейрогенный мочевой пузырь.
Для оценки степени нарушения когнитивных функций использовались шкала MMSE и тест рисования часов. Получены результаты 28 и 10 баллов соответственно.
Проводилась дифференциальная диагностика между болезнью Паркинсона, паркинсонизмом «плюс», прогрессирующим надъядерным параличом, кортикобазальной дегенерацией, нормотензивной гидроцефалией, вторичными дисметаболическими энцефалопатиями, мультисистемной атрофией.
На основании клинической картины, характера течения заболевания, данных дополнительных исследований, вышеприведенных критериев диагностики пришли к заключению, что у пациента мультисистемная атрофия мозга с выраженным акинетикоригидным и атактическим синдромом, рефлекторнопирамидной недостаточностью в конечностях, вегетативной недостаточностью (пандизавтономией), выраженными нарушениями передвижения и самообслуживания.
Получал лечение: левоком 600 мг/сутки в 3 приема, неомидантан внутрь по 100 мг 2 раза в сутки, кортексин 2,0 в/м, цитофлавин 10,0 в/в капельно на 200,0 физиологического раствора № 7, затем внутрь по 1 таблетке 2 раза в сутки беллатаминал внуть по 1 таблетке 3 раза в день, кудесан Q10 по 10 капель 2 раза в сутки во время еды, семакс 0,1% раствор по 2 капли в каждый носовой ход 2 раза в день.
Справка: мультисистемная атрофия — спорадическое прогрессирующее нейродегенеративное заболевание с поражением базальных ганглиев, ствола мозга, мозжечка, спинного мозга, проявляющееся паркинсонизмом, мозжечковой атаксией, вегетативной недостаточностью и пирамидным синдромом в различных сочетаниях. Мультисистемная атрофия — самостоятельная нозологическая форма, являющаяся одним из вариантов мультисистемных дегенераций.
В зависимости от преобладания тех или иных синдромов выделяют 3 основных клинических типа МСА:
1) стриатонигральную дегенерацию (стриатонигральный тип), характеризующуюся преобладанием в клинической картине симптомов паркинсонизма;
2) оливопонтоцеребеллярную атрофию (оливопонтоцеребеллярный тип), характеризующуюся преобладанием в клинической картине мозжечковой атаксии;
3) синдром Шая — Дрейджера, характеризующийся доминированием в клинической картине симптомов прогрессирующей вегетативной недостаточности, прежде всего ортостатической гипотензии.
Критерии диагностики мультисистемной атрофии (Gilman et al., 1998)
Клинические проявления (характерные для заболевания):
Вегетативная/тазовая дисфункция (ортостатическая гипотензия со снижением систолического давления не менее чем на 20 мм рт.ст. или диастолического — не менее чем на 10 мм рт.ст. в течение 3 мин стояния; недержание мочи и неполное опорожнение мочевого пузыря).
Паркинсонизм (гипокинезия; ригидность; постуральная неустойчивость, не связанная с первичным нарушением зрения, проприоцепции, вестибулярных или мозжечковых функций; дрожание покоя и/или постуральное дрожание).
Мозжечковая атаксия (статолокомоторная атаксия с увеличением площади опоры, неравномерными по длине и направлению шагами; скандированная речь; дискоординация конечностей; нистагм).
Пирамидный синдром (оживление сухожильных рефлексов с наличием разгибательных стопных знаков).
Критерии, удостоверяющие диагноз:
Вегетативная/тазовая дисфункция (ортостатическая гипотензия со снижением систолического давления не менее чем на 30 мм рт.ст., диастолического — не менее чем на 15 мм рт.ст. через 3 мин стояния и/или недержание мочи и нарушение эрекции у мужчин).
Паркинсонизм (гипокинезия в сочетании не менее чем с одним другим паркинсоническим симптомом).
Мозжечковая атаксия (статолокомоторная атаксия в сочетании не менее чем с одним другим мозжечковым симптомом).
Критерии, исключающие диагноз:
Начало в возрасте до 30 лет; положительный семейный анамнез; наличие анамнестических, клинических или параклинических признаков иного заболевания, способного вызвать аналогичные симптомы; галлюцинации, не связанные с приемом лекарственных средств; наличие деменции; резкое замедление вертикальных саккад или паралич вертикального взора; признаки нарушения корковых функций (афазия, синдром «чужой» руки, дисфункция теменной коры).
В настоящее время не существует эффективного лечения мозжечковых нарушений при МСА, поэтому фармакотерапия в значительной степени направлена на облегчение симптомов паркинсонизма и дисавтономии. Леводопа (плюс ингибитор ДОФАдекарбоксилазы) в дозе до 1000 мг/сут при условии хорошей переносимости. Агонисты дофаминовых рецепторов являются препаратами второй линии (дозы такие же, как и при болезни Паркинсона), амантадин — препарат третьей линии (100 мг до 3 раз/сут), семакс 0,1% раствор капли в нос. Терапия ортостатической гипотензии часто достаточно сложная, вместе с тем она улучшает качество жизни больных МСА. Низкое артериальное давление может не сопровождаться какойлибо симптоматикой, вероятно, изза того, что мозговой кровоток у них поддерживается на адекватном уровне за счет механизмов ауторегуляции даже при снижении систолического артериального давления до 60 мм рт.ст. Когда данное состояние создает дискомфорт, его можно избежать путем ограничения действия провоцирующих факторов — большого количества пищи, алкоголя, физических нагрузок, внешнего теплового воздействия. К немедикаментозным стратегиям относятся ношение эластичных колготок, возвышенное положение головного конца кровати на ночь, увеличение содержания соли в пищевом рационе. Урологические проблемы при МСА вызваны сочетанием центральных и периферических неврологических нарушений, которые иногда наслаиваются на местные патологические изменения, например гипертрофию простаты и слабость мышц промежности. Холинолитики периферического действия эффективны при недержании мочи, однако часто индуцируют ее задержку; десмопрессин, принятый на ночь, обеспечивает регресс никтурии. При неполном опорожнении мочевого пузыря необходима периодическая самокатетеризация. Поскольку результаты фармакотерапии МСА в целом неблагоприятные, важной является роль и других терапевтических стратегий. Физиотерапия помогает поддерживать двигательную активность и предотвращает контрактуры, логопедия улучшает артикуляцию, коммуникативные способности и облегчает глотание, трудотерапия способствует преодолению ограничений, связанных с необратимой инвалидизацией, психотерапия обеспечивает эмоциональную поддержку как пациенту, так и семье. Дисфагия часто сопровождается необходимостью кормления через назогастральный зонд и даже применения чрескожной эндоскопической гастростомии. Обеспечение больного инвалидной коляской необходимо в связи со склонностью к частым падениям и атаксией походки.
В настоящее время улучшилось понимание клинических проявлений МСА [6–8]. Этого нельзя сказать о лечении, результаты которого умеренные или вообще отсутствуют. Поэтому существует крайняя необходимость в разработке будущих терапевтических исследований, направленных на изучение новых симптоматических и нейропротекторных препаратов, а также оптимизацию немедикаментозных вмешательств при данной патологии.
Методы диагностики
Обследоваться надо у невролога. Для постановки диагноза нужно динамическое наблюдение пациента с применением церебрального МРТ. В случае противопоказаний проводят компьютерную томографию ПЭТ, ОФЭКТ.
В начале развития МСА МРТ не покажет атрофических изменений мозговой ткани, но поможет исключить опухоль, энцефалит, рассеянный склероз. Через 1―3 года интенсивной прогрессиивыявляют расширение IV желудочка, выраженную дегенерацию подкорковых ганглий, нижней половины моста, мозжечка, скорлупы.
На осмотре невролог оценивает наличие вегетативной недостаточности в комбинации спаркинсонизмоми/или дисфункцией мозжечка.
Мультисистемная атрофия не подтверждается, если:
- МСА начала развиваться до 30 либо после 75 лет;
- вегетативная недостаточность не сочетается ни с мозжечковой дисфункцией, ни с паркинсонизмом;
- патология также есть у близких родственников (семейный анамнез);
- у пациента выявлена деменция, признаки похожей на МСА болезни;
- лечение паркинсонизма эффективно лекарствами леводопы.
Для постановки диагноза ортостатической пробой исследуют функции вегетативной нервной системы. Нарушение работы тазовых структур выявляют при проведении электромиографии сфинктеров.
На развитие мультисистемной атрофии указывает наличие:
- ортостатической гипотензии — снижение давления после принятия вертикального положения;
- нерегулярного тремора;
- холодности стоп и кистей, скованности их сочленений;
- храпа, который вновь появился либо усилился;
- плача или смеха, несоответствующих переживаемой эмоции;
- тяжелых нарушений речи, голоса — дизартрия, дисфония;
- затрудненных вдохов — инспираторная одышка;
- недержания мочи;
- нарушений эректильной функции;
- кривошеи с наклоном головы к груди — антероколлис;
- непроизвольных движений мимических мышц, языка — орофациальная дистония;
- искривления позвоночника в грудопоясничном переходе с наклонением туловища вперед — камптокормия;
- учащения случаев падения;
- мозжечкового синдрома/паркинсонизма + вегетативной недостаточности.
Справка! МСА достоверно подтверждается патоморфологическим исследованием нейроглии. При жизни биоматериал с глиальными клетками получают посредством биопсии мозга или ткань изымает патологоанатом во время вскрытия.
Паркинсонизм: клиника, диагноз и дифференциальный диагноз
ММА имени И.М. Сеченова
Клиническая картина
Паркинсонизм (П) – синдром, связанный с поражением базальных ганглиев и их связей. Его главные проявления – бедность движений (акинезия) и повышение тонуса мышц (ригидность). Поэтому П по–другому называют акинетико-ригидным синдромом
. В прямом понимании термин
«акинезия»
означает отсутствие движений или их обеднение. Акинезия проявляется отсутствием так называемых ассоциированных, спонтанных движений – например, во время ходьбы отсутствует физиологическая синкинезия в виде раскачивания рук или амплитуда этого движения резко уменьшена (ахейрокинез); обеднена мимика – отмечается редкое моргание, уменьшены эмоциональные движения (гипомимия). В понятие акинезии входит и затруднение начала (инициации) движений. При П имеет место также замедленность произвольных двигательных актов и их истощаемость, что некоторые исследователи обозначают термином «брадикинезия». Например, при повторном сжимании и разжимании кисти, пронации-супинации рук, постукивании ногой по полу и т. д. заметно, что амплитуда и сила первых движений больше, чем последующих. Помимо акинезии, обеднение движений обозначают таким же по смыслу термином – «гипокинезия» (а синдром П – гипокинетико-ригидным синдромом).
Ригидность
– это особый тип повышения тонуса мышц. При пассивных движениях в суставах (сгибание–разгибание, пронация–супинация и т.д.) исследователь чувствует постоянное сопротивление, одинаковое на протяжении всего движения. Это ощущение похоже на то, которое возникает при сгибании и разгибании свинцовой трубки (в отличие от другого вида повышения тонуса – спастичности, при котором наибольшее сопротивление пассивным движениям отмечается в начале движения. Последнее называют «феноменом складного ножа»). Ригидность при П опеределяется как в мышцах шеи и туловища, так и в мышцах конечностей. Нередко повышение тонуса преобладает в мышцах–сгибателях, из-за чего формируется согбенная поза туловища с несколько согнутыми в локтевых суставах и приведенными к туловищу руками и согнутыми в коленных суставах ногами. Эту позицию называют «позой сгибателей» или «позой просителя».
Еще одним характерным симптомом является феномен «зубчатого колеса
». Он встречается наряду с ригидностью, но может иметься и в ее отсутствие. Во время сгибания и разгибания в суставах исследователь ощущает неравномерное сопротивление производимому движению, создающее впечатление соскальзывания зацепляющихся между собой зубцов шестерен.
В дебюте П многие больные не замечают за собой каких-либо нарушений. Часто родственники и друзья пациента впервые обращают внимание на уменьшение выразительности лица (часто интерпретируя это, как проявление депрессии), ахейрокинез и замедленность движений, особенно при одевании, еде и ходьбе. Впоследствии сами больные отмечают уменьшение ловкости рук, особенно при выполнении тонких движений, таких как застегивание пуговиц, завязывание шнурков. Нарушаются привычные виды деятельности, требующие проворности рук (например, игра на музыкальных инструментах, работа на печатной машинке или за компьютером). Возникают нарушение письма – почерк меняется, становится мелким (микрография), менее разборчивым, а на продвинутых стадиях вовсе непонятным; трудности при чистке зубов, бритье и т.д. Значительно затруднены повороты в постели, проблемой является вставание с кресла или низкого стула, зачастую пациенты не в состоянии лечь в ванну и встать из нее, поэтому могут мыться только под душем. Многие больные связывают эти явления со «слабостью» и предъявляют соответствующие жалобы. Обычной жалобой является также отсутствие энергии и быстрая утомляемость. Впоследствии становится затруднен прием пищи из-за трудности жевания, возникает поперхивание при глотании. Из-за акинезии мыщц глотки глотательные движения становятся более редкими, что обусловливает развитие слюнотечения. Последнее может быть столь интенсивным, что больные вынуждены постоянно пользоваться платком или полотенцем.
Часто акинезия и ригидность сочетаются с тремором
. Типичный паркинсонический тремор очень своеобразен, его трудно перепутать с другим видом дрожания. Во-первых, в преобладающем большинстве случаев его отмечают в руках, реже – в ногах. Следует помнить, что паркинсоническое дрожание никогда не начинается с головы, и даже на очень продвинутых стадиях П дрожание головы – исключительная редкость. Дрожание нижней челюсти и языка, напротив, можно наблюдать достаточно часто (обычно на поздних этапах). Во-вторых, паркинсонический тремор является тремором покоя. Это значит, что он наблюдается (или максимально выражен) в ситуации, когда руки пациента в расслабленном состоянии лежат на его коленях, ручках кресла, столе и т.д. При совершении движений дрожание исчезает или становится гораздо менее интенсивным. В-третьих, при паркинсоническом дрожании кисти пациента совершают круговые движения, а большой и указательный пальцы при этом двигаются навстречу друг другу. Это напоминает движения, совершаемые при счете денег или скручивании бумажных шариков. Поэтому паркинсоническое дрожание образно называют тремором по типу «счета монет» или «скатывания пилюль». Следует помнить, что дрожание не является обязательной составляющей синдрома П.
Болезнь Паркинсона. На рисунке А показан участок мозга здорового человека с хорошо пигментированным черным веществом. На рисунке В — участок мозга пациента, страдающего болезнью Паркинсона. Заметно отсутствие пигментации черного вещества.
П сопровождается также
нарушением равновесия вследствие расстройства
так называемых постуральных рефлексов. Последние обеспечивают прямостояние, сохранение устойчивости при разных движениях (движения рук, ходьба, присаживание и вставание, наклоны и т. д.), а также при внезапных дестабилизирующих воздействиях (например, при толчке, спотыкании и др.). Расстройство постуральных рефлексов проявляется в частых падениях больных. При более легких нарушениях падений не отмечается, но имеет место неустойчивость при легком толчке в грудь или спину. При присаживании на стул больные падают на него, нередко резко откидываясь на спинку. Постуральные нарушения обусловливают также возникновение
пропульсий –
одной из характерных черт П. Пропульсии выражаются в том, что при ходьбе больного «тянет вперед», он короткими шагами быстро идет вперед, как бы «догоняя свой центр тяжести», и не в состоянии резко остановиться. Нередко это заканчивается падением.
П – это синдром, который может встречаться в рамках разных заболеваний. Наиболее часто причиной П является идиопатический паркинсонизм, или болезнь Паркинсона (БП)
. П часто наблюдается в рамках других идиопатических дегенеративных заболеваний нервной системы. Последние часто называют паркинсонизмом–плюс. К этой группе относятся мультисистемная атрофия, прогрессирующий надъядерный паралич, болезнь диффузных телец Леви, кортико-базальная дегенерация. Нередко встречается
симптоматический П (не связанный с первичным дегенеративным заболеванием нервной системы)
. К нему относят лекарственный, сосудистый, посттравматический, постэнцефалитический, токсический П, П при опухолях головного мозга и гидроцефалии.
П характеризует также клиническую картину некоторых наследственных дегенеративных заболеваний ЦНС
. К их числу относятся гепатолентикулярная дегенерация (болезнь Вильсона-Коновалова), спиноцеребеллярные атаксии, семейная кальцификация базальных ганглиев, болезнь Гентингтона и др. Таким образом, синдром П не является синонимом БП.
Диагноз и дифференциальный диагноз
Эссенциальный тремор
На первом этапе диагностической работы необходимо убедиться, что у больного действительно имеется синдром П. Наиболее часто за истинный П ошибочно принимают эссенциальный тремор
(ЭТ) и своеобразные изменения походки, связанные с сосудистой патологией головного мозга. Таким пациентам очень часто неправильно ставят диагноз БП.
ЭТ – наследственное заболевание (хотя нередки и спорадические случаи), которое характеризуется двусторонним, симметричным тремором рук. В отличие от паркинсонического тремора дрожание рук при ЭТ отсутствует в покое и возникает при работе мышц. Обычно оно максимально выражено тогда, когда больной вытягивает руки перед собой, может присутствовать также при совершении произвольных движений, часто затрудняя бытовые виды деятельности (например, еду). Обычно при ЭТ наблюдается тремор головы по типу «нет-нет» или «да-да» и тремор голоса. Последние могут быть первыми проявлениями ЭТ, а также встречаться изолированно в отсутствие дрожания конечностей. В отличие от ЭТ дрожание головы и голоса, как правило, не встречаются при БП. Напротив, вертикальный тремор нижней челюсти в состоянии покоя и дрожание ног часто имеют место при БП и редко при ЭТ. Характерным для ЭТ признаком, который имеет диагностическое значение, является исчезновение тремора под воздействием алкоголя.
Хорошо влияют на ЭТ также b-блокаторы. При ЭТ может иметь место феномен «зубчатого колеса», но в отличие от БП никогда не отмечается истинной акинезии и ригидности мышц. ЭТ чаще страдают пожилые люди, хотя он может встречаться и среди очень молодых людей.
В прошлом сосудистое поражение головного мозга считалось самой частой причиной П и даже рассматривалось, как причина БП. В настоящее время стало ясно, что БП – первичное дегенеративное заболевание ЦНС и сосудистое поражение не играет этиологической роли в ее развитии. Цереброваскулярные заболевания очень редко вызывают истинный П, что послужило поводом ввести понятие «атеросклеротический псевдопаркинсонизм», или «сосудистый псевдопаркинсонизм». Используются также такие обозначения, как «паркинсонизм нижней половины тела» и «паркинсоническая атаксия». Пациенты, страдающие этими нарушениями, как правило, пожилые люди, часто имеющие в анамнезе артериальную гипертензию. Наиболее характерной чертой сосудистого псевдопаркинсонизма является выраженное нарушение походки:
больные ступают короткими шагами, нередко широко расставляя ноги (что нехарактерно для истинного П). Часто затруднена инициация ходьбы и повороты, имеют место также нарушение равновесия и падения. Наряду с этим в верхней половине тела, руках и лице полностью отсутствуют какие-либо проявления П: мимика больных живая, голос нормально модулирован, движения рук свободны, ахейрокинез отсутствует. Тремор также не является частью клинической картины. Сосудистый псевдопаркинсонизм может сочетаться со снижением высших психических функций, пирамидными знаками (хотя это не обязательное явление). Всем больным с подобной клинической картиной необходимо проведение визуализации головного мозга –
магнитно-резонансной томографии
(МРТ) или
компьютерной томографии
(КТ).
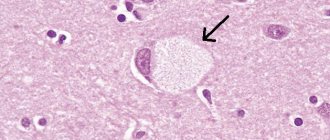
Тельца Леви при болезни Паркинсона. В цитоплазме нейрона определяется эозинофильное ядро окруженное неокрашенной зоной.
Последние, как правило, выявляют множественные мелкие очаги сосудистого происхождения (лакуны) в области базальных ганглиев и/или изменения белого вещества полушарий головного мозга (лейкоареоз). Следует иметь в виду, что схожая клиническая картина может возникать иногда при нормотензивной гидроцефалии и реже – при опухолевом поражении головного мозга. При нормотензивной гидроцефалии наряду с нарушением ходьбы по типу описанного выше имеют место деменция и расстройство функции тазовых органов (эту триаду симптомов называют
триадой Хакима–Адамса
). МРТ головы, как правило, обнаруживает резко расширенные боковые желудочки.
Болезнь Паркинсона
Самой частой причиной истинного П (более чем 80%), как говорилось выше, является БП. Это дегенеративное заболевание, при котором основной патологический процесс развивается в нигро-стриарной системе. В частности, прогрессирующей дегенерации подвергаются нейроны черной субстанции, которые вырабатывают дофамин. В результате развивается дефицит дофамина, что и обусловливает клиническую симптоматику. БП обычно страдают люди пожилого возраста. Все описанные выше симптомы П при БП развиваются в полной мере. Для БП типично асимметричное начало.
В дебюте болезни симптомы касаются, как правило, одной стороны тела, возникая обычно в первую очередь в руке (чаще в правой), затем в ноге на той же стороне. Нередко первое проявление паркинсонизма в руке – микрография и ахейрокинез. Стадия гемипаркинсонизма длится обычно несколько месяцев, редко несколько лет. Затем симптоматика вовлекает контралатеральные конечности, а также мышцы туловища и шеи. Симптомы П могут возникать постепенно (например, изначально появляется акинезия и ригидность в руке, затем присоединяется дрожание). Может быть и обратная ситуация. Следует помнить, что тремор отсутствует у 30% больных БП в начале болезни, а в некоторых случаях в течение всей болезни. В связи с этим
выделяют разные формы БП
– акинетико-ригидную, ригидно-дрожательную и дрожательную. На стадии гемипаркинсонизма рука больного несколько согнута в локтевом суставе, не раскачивается во время ходьбы и как бы прижата к туловищу. На более продвинутых стадиях, когда такие же изменения возникают и на другой стороне, а ригидность и акинезия вовлекают мышцы туловища и шеи, формируется специфическая паркинсоническая
«поза просителя»
. Нарушение равновесия и падения возникают на продвинутых стадиях БП, обычно через несколько лет после начала болезни. В рамки БП не входят мозжечковые и пирамидные расстройства, хотя имеется тенденция к оживлению глубоких рефлексов на более пораженной стороне. Иногда может наблюдаться экстензорная установка стопы и пальцев, напоминающая симптом Бабинского, так называемая стриарная стопа. Симптомы БП, как правило, значительно уменьшаются под воздействием препаратов
леводопы
. Если паркинсонические симптомы не реагируют или слабо реагируют на прием достаточной дозы этих препаратов, диагноз БП следует подвергнуть сомнению. При длительном лечении лекарствами, содержащими леводопу, практически у всех больных возникают побочные явления в виде двигательных флуктуаций и дискинезий. Последние выражаются в следующем: до очередного приема препарата отмечается выраженное нарастание симптомов П, нередко до полной обездвиженности больного, а в период, когда концентрация препарата в крови достигает максимальных значений, акинезия и ригидность полностью отсутствуют (феномен «включения и выключения») и даже наблюдаются избыточные непроизвольные движения (дискинезии). Последние иногда настолько выражены, что при первом взгляде на пациента создается впечатление, что он болен хореей.
Диагноз БП – это клинический диагноз.
Параклинические методы исследования обычно выявляют неспецифические изменения. Нередко у больных с классической БП в головном мозге имеются сосудистые изменения. Это, однако, ни в коем случае не является свидетельством сосудистого генеза болезни. Просто у большинства пожилых людей, в том числе практически здоровых, подобные изменения могут иметь место. Тем не менее, в некоторых случаях на БП наслаивается атеросклеротический псевдопаркинсонизм, что может быть причиной необычной для БП походки и ранних постуральных нарушений.
Лекарственный паркинсонизм
Лекарственный П может быть обусловлен препаратами, которые воздействуют на пресинаптические дофаминовые нейроны черной субстанции, истощая запасы дофамина в них (например, резерпин) или, наиболее часто, нейролептиками, которые блокируют постсинаптические дофаминовые рецепторы, такими как производные фенотиазина (хлорпромазин), бутирофеноны (галоперидол), тиоксантины (флупентиксол) и бензамиды (сульпирид). Эти препараты часто применяются при психических заболеваниях. Также П может быть вызван прохлорперазином (используется при рвоте, головокружении и неустойчивости), метоклопрамидом (применяется при заболеваниях желудочно-кишечного тракта, для купирования тошноты и рвоты). П может быть обусловлен также циннаризином, который является атипичным блокатором кальциевых каналов (применяется при вестибулярных расстройствах). Комбинация нейролептиков и антидепрессантов также может быть причиной П.
В психиатрических лечебницах лекарственный П встречается часто. Гипомимия и ахейрокинез – настолько обычное явление среди этого контингента больных, что психиатры часто даже не обращают на них внимание. Тремор встречается менее часто, но может иметь вид классического паркинсонического дрожания. Более того, лекарственный П может быть асимметричным, подобно БП, и нередко совершенно не отличим от БП. Одним из признаков, по которому следует заподозрить лекарственный П, является наличие наряду с акинетико-ригидным синдромом насильственных движений в виде, например, оро-мандибулярной дискинезии (непроизвольные жевательные и/или сосательные движения) или дистонических явлений (спастической кривошеи, окулогирных кризов), стереотипий, акатизии (неусидчивость). Нередко тяжелый лекарственный П сопровождается выраженной дизартрией и дисфагией. Индивидуальная чувствительность к дофаминблокирующим препаратам очень разнообразна. Некоторые пациенты без каких-либо проблем переносят длительное лечение большими дозами этих веществ, тогда как у других побочные явления развиваются уже на малых дозах. Чаще
, однако,
признаки лекарственного П возникают при приеме больших доз нейролептиков
. Лекарственный П обычно развивается постепенно в течение дней или недель. У большинства больных первые признаки появляются через 3 нед после начала лечения. Наиболее часто встречающиеся дебютные признаки – гипомимия и недостаточное раскачивание рук во время ходьбы.
Течение лекарственного П может быть различным. В большинстве случаев он постепенно, в течение нескольких недель, а иногда и дней, проходит после прекращения приема вызвавшего его препарата. Тем не менее нередки случаи, когда П длится в течение месяцев, иногда почти год. Такая ситуация наблюдается при применении нейролептических препаратов, способных к депонированию. В редких случаях лекарственный П не проходит и продолжает прогрессировать, несмотря на прекращение приема вызвавшего его агента. Подобные случаи чаще встречаются среди пожилых людей. Считается, что в таких случаях прогрессирует не сам по себе лекарственный П, а начинает развиваться БП.
Мультисистемная атрофия
Мультисистемная атрофия (МСА) – это спорадическое заболевание, возникающее у взрослых лиц, при котором в отличие от БП дегенерации подвергается не только нигро-стриарная система, но также множество других образований ЦНС, включая мозжечок и его связи, пирамидные пути и образования вегетативной нервной системы (отсюда и происходит название болезни). Соответственно клинически МСА характеризуется сочетанием П, мозжечковых нарушений, пирамидных расстройств и прогрессирующей вегетативной недостаточности
(ПВН). П при МСА обусловлен не только поражением клеток черной субстанции, что вызывает дефицит дофамина, но также дегенерацией тех постсинаптических рецепторов, с которыми должен взаимодействовать дофамин.
МСА наиболее часто дебютирует после 50 лет и в отличие от БП значительно укорачивает продолжительность жизни больных, становясь причиной смерти в среднем в течение 9 лет от появления первых симптомов. П имеет место у 90% больных МСА, а доминирующим клиническим признаком является у 80%. Мозжечковые и пирамидные расстройства проявляются у 50% пациентов. Практически у всех больных отмечаются признаки ПВН. Вегетативные расстройства встречаются также при БП, поэтому одним из ключевых моментов дифференциальной диагностики МСА и БП является выяснение того, когда возникли признаки ПВН. При МСА они часто возникают еще до начала двигательных проявлений, нередко опережая последние на несколько лет, тогда как при БП они встречаются редко и, как правило, через несколько лет после начала болезни. У мужчин часто первым симптомом становится импотенция. Обычное явление как среди мужчин, так и среди женщин – недержание мочи. Нередко из-за этого больные, до того как обратиться к неврологу, попадают на прием к урологу и даже подвергаются ненужному хирургическому вмешательству. Для сравнения: при БП имеет место лишь учащение мочеиспускания и иногда императивные позывы к опорожнению пузыря в результате гиперрефлексии мышцы-детрузора. Для большинства лиц с МСА характерна также ортостатическая гипотензия (ОГ), которая может быть причиной липотимических состояний и обмороков. У больных с БП ОГ также встречается, но менее часто и, как правило, на продвинутых стадиях болезни. Характерна зябкость конечностей – так называемый симптом холодных рук. Примерно у 13% больных МСА имеет место выраженный респираторный стридор из-за паралича абдуктора голосовых связок. Стридор вначале отмечается только во время сна и становится причиной очень громкого храпа, а по мере прогрессирования проявляется и во время бодрствования. Паралич абдуктора голосовых связок настолько патогномоничен для МСА, что, по мнению некоторых исследователей, при сочетании П и недавно возникшего выраженного ночного храпа, следует подозревать МСА. Сам синдром П у больных МСА развивается не так, как при БП. В частности, при МСА П, обычно, начинается с общей замедленности движений, которая затрагивает руки и ноги как с левой, так и с правой стороны. Хотя симптоматика также бывает асимметрична, стадия гемипаркинсонзма в отличие от БП, как правило, отсутствует. Явления П при МСА могут начинаться с дизартрии — артикуляция больного становится более медленной, слова менее разборчивы и произносятся менее четко (смазанность речи). БП практически никогда не начинается с дизартрии. Часто у больных МСА присутствует выраженная дисфония – голос очень глухой и прерывающийся, нередко это сочетается с дисфагией, что становится причиной частых поперхиваний во время еды. Тремор покоя также может иметь место при МСА, однако классический вариант паркинсонического дрожания, описанный выше, по типу «счета монет», наблюдается лишь в 10% случаев. Напротив, часто имеет место постуральное дрожание и тремор во время движения. Нередко как тремор ошибочно расценивают нерегулярные миоклонические подергивания пальцев, характерные для МСА. Для этой болезни характерны также чувствительные к стимулам миоклонии. У ряда больных развивается так называемый диспропорциональный антеколлис
– голова значительно наклонена вперед, несмотря на то, что в целом «согбенная поза» выражена незначительно. Этот признак, однако, не считается специфическим для МСА. Следует помнить, что деменция не является частью клинической картины МСА. В целом МСА протекает более злокачественно, чем БП, и значительно инвалидизирует больных нередко уже в течение первого года болезни.
В клинической картине МСА может преобладать та или иная симптоматика. Те случаи, при которых на первый план выступает П, обозначают термином нигро-стриальная дегенерация
(СНД); если в клинической картине ведущим является мозжечковый синдром, это состояние называют
оливо-понто-церебеллярной атрофией
(ОПЦА); случаи, когда ядром клинической картины является ПВН, обозначают эпонимическим названием –
синдром Шая–Дрейджера
(СШД).
Несмотря на все различия, нередки ситуации, когда МСА невозможно отличить от БП. В таких случаях следует в основном ориентироваться на эффект препаратов леводопы. При БП эти препараты оказывают драматический положительный эффект, тогда как при МСА этот эффект не столь выражен, кратковременен, а нередко отсутствует совсем. Это обусловлено поражением постсинаптических рецепторов, с которыми должна взаимодействовать леводопа.
Признаки, при наличии которых у больных с П следует подумать об МСА
, следующие: быстрое прогрессирование симптоматики; рано развившиеся нарушения равновесия и падения; отсутствие улучшения при лечении препаратами леводопы или недостаточный эффект этих препаратов; ПВН; пирамидные и/или мозжечковые знаки; похолодание, зябкость конечностей; контрактуры; диспропорциональный антеколлис; выраженная дисфония, дисфагия или дизартрия; респираторный стридор; нерегулярный тремор или миоклонии.
Из параклинических методов исследования в диагностике МСА могут помочь специальные тесты на выявление ПВН, электромиография наружного сфинктера мочеточника или анального сфинктера,
которая выявляет денервационные изменения в них из-за поражения сакрального ядра Онуфа, откуда иннервируются эти мышцы. МРТ головного мозга нередко выявляет атрофию мозжечка, а при высокой разрешающей способности метода (с мощностью магнитного поля 1,5 тесла) в Т2 взвешенном изображении определяется гиперинтенсивный сигнал щелевой формы по наружному краю скорлупы. Однако ни одно из перечисленных изменений, взятое в отдельности, не является специфичным только для МСА, поэтому их следует оценивать только вкупе с клинической картиной. В целом диагноз МСА, так же как и БП, в основном является клиническим диагнозом. При жизни пациента диагностируется только «возможная» или «вероятная» МСА на основе специальных диагностических критериев, а достоверная диагностика возможна только при патоморфологическом обследовании в случае обнаружения специфических гистологических маркеров болезни – глиальных цитоплазматических включений.
Прогрессирующий надъядерный паралич
Прогрессирующий надъядерный паралич (ПНП), или синдром Стила–Ричардсона–Ольшевского
, – еще один вариант дегенераций, поражающих множественные отделы нервной системы, при котором нередко ошибочно ставится диагноз БП. ПНП начинается позже, чем БП и МСА, – чаще после 70 лет. Средняя продолжительность жизни после появления первых симптомов составляет 6–7 лет. При этой болезни П характеризуется симметричностью, акинезия и ригидность в конечностях выражены не столь ярко, иногда практически отсутствуют, а страдает в основном аксиальная мускулатура, т.е. мышцы туловища и шеи. Нередко у больных ПНП отмечается несколько закинутая назад голова, хотя наличие этого признака необязательно, голова может также быть наклонена вперед или находиться в нормальном положении.
Первым симптомом этого заболевания в большинстве случаев является нарушение равновесия и падения.
Нередко ПНП дебютирует с дизартрии, наряду с которой могут иметь место непроизвольные глубокие вдохи, похожие на стон. Клиническим маркером болезни является
надъядерная офтальмоплегия
(НО), или надъядерный парез взора, вследствие поражения специфических образований среднего мозга. НО можно диагностировать, когда больной не в состоянии произвольно изменить направление взора, тогда как синкинетические и рефлекторные движения глаз сохранены. Например, при ПНП больной не может произвольно переместить глазные яблоки вверх и/или вниз, но при этом сохранен
феноменБелла
– отведение глазных яблок вверх при закрывании глаз;
окулоцефалический рефлекс
(при фиксированном на одной точке взоре во время поворота или наклона головы в какую-либо сторону глазные яблоки содружественно отводятся в противоположную). НО редко развивается в дебюте ПНП, обычно она присоединяется к другим симптомам в среднем через 2–4 года. В начале же болезни может иметь место выраженное замедление движений глаз, своеобразный застывший взор, жжение глаз и ощущение «песка в глазах» из-за очень редкого моргания характерного для ПНП. Часто отмечается непроизвольное насильственное зажмуривание глаз (блефароспазм), особенно при воздействии яркого света или других стимулов. Из-за этого некоторые больные вынуждены постоянно носить солнцезащитные очки. Может наблюдаться также так называемая
апраксия открывания век
, которая проявляется в том, что больному трудно открыть закрытые (не обязательно зажмуренные) глаза. Из-за нарушения движений глазных яблок пациентам бывает трудно есть, так как они не видят полностью расположенную перед ними тарелку. Нередко они роняют себе пищу на грудь («симптом грязного галстука»). Им трудно спускаться по лестнице, садясь на стул, они садятся мимо сиденья. Все это в сочетании с застывшим взором и редким морганием часто производит ошибочное впечатление, что больной неадекватен, неправильно ведет себя, малодоступен контакту. Из-за этого пациенты с ПНП могут иногда попадать на прием к психиатру. Для ПНП характерны также выраженные псевдобульбарные расстройства в виде оживления лицевых аксиальных рефлексов, у больных нередко отмечается выраженное углубление складок на лице (носогубных, лобных). Характерно также выраженное замедление психических процессов, из-за чего нередко больные долго думают перед ответом даже на элементарные вопросы.
Таким образом, хотя ПНП поверхностно схожа с БП из-за наличия ригидности аксиальных мышц, выраженной гипомимии и нарушения ходьбы, при внимательном осмотре обнаруживается, что в дистальных отделах конечностей П практически отсутствует или выражен очень мягко. Практически никогда не встречается классический тремор покоя. Препараты леводопы обычно не эффективны. Вегетативная недостаточность не характерна для ПНП. МРТ головного мозга может выявлять атрофию среднего мозга, однако это опять же является неспецифическим признаком и диагноз ПНП в основном базируется на клинических проявлениях. Так же, как в случае с МСА, клинически с помощью специально разработанных критериев ставится диагноз «возможного» или «вероятного» ПНП, а достоверный диагноз ставится при патоморфологическом исследовании в случае обнаружения специфических маркеров (нейрофибриллярных клубочков).
Болезнь диффузных телец Леви
В последнее время стали выделять новую нозологическую форму, протекающую с синдромом П – болезнь диффузных телец Леви (БДТЛ). Тельца Леви – это внутриклеточные эозинофильные цитоплазматические включения, которые обнаруживаются в клетках черного вещества при БП и считаются маркером этой болезни. При БДТЛ они встречаются не только в черном веществе, но в большом количестве широко диссеминированы по всему головному мозгу. Диагноз БДТЛ – патоморфологический диагноз.
Клинически это заболевание характеризуется П, который обычно хорошо лечится препаратами леводопы, наряду с деменцией с выраженными зрительными галлюцинациями. Типичным является также
флуктуация выраженности расстройств высших психических функций
– в основном за счет изменения способности концентрации внимания.
Гепатолентикулярная дегенерация (болезнь Вильсона-Коновалова) и другие заболевания
Существует несколько более редких причин истинного или псевдопаркинсонизма. Одна из них, о которой следует всегда вспомнить при наличии П у людей моложе 45 лет (в том числе у детей), это гепатолентикулярная дегенерация, или болезнь Вильсона-Коновалова
. Это наследственное заболевание, при котором отмечается нарушение метаболизма меди в организме из-за недостаточности фермента церулоплазмина. В результате медь в избыточном количестве откладывается в печени, базальных ганглиях и вокруг радужной оболочки глаза. Болезнь Вильсона-Коновалова следует подозревать не только при наличии П у молодых людей, но и при возникновении у них других признаков поражения экстрапирамидной системы (например, дистонии) или психических расстройств. Диагностика основана на обнаружении с помощью щелевой лампы отложения меди вокруг радужки –
кольцо Кайзера–Флейшера
. Последнее на стадии неврологических проявлений имеет место у 98% больных. Диагностическое значение имеет также
исследование экскреции меди с мочой и концентрации церулоплазмина в крови
. Последнее, однако, в отсутствие кольца Кайзера–Флейшера и нормальной экскреции меди не имеет диагностической значимости. Если ситуация продолжает оставаться неясной, проводится биопсия печени или генетическое тестирование. Болезнь Вильсона-Коновалова довольно успешно лечится с помощью D-пеницилламина и препаратов цинка в сочетании с диетой.
Токсический П
может иметь место при интоксикации марганцем. Нередко встречается П, вызванный токсином МФТП (1-метил-4-фенил- 1, 2, 3, 6-тетрагидропиридин), так как оно избирательно поражает черную субстанцию, вызывая гибель дофаминовых нейронов. Схожий с этим веществом токсин вырабатывается при кустарном приготовлении некоторых наркотических веществ, поэтому П нередко наблюдается среди молодых людей, страдающих наркоманией. Наряду с П при этом могут отмечаться пирамидные и псевдобульбарные симптомы.
П встречается при энцефалопатии боксеров
. Считается, что в таком случае этиологическую роль играют регулярно получаемые множественные черепно-мозговые травмы. Следует помнить, что энцефалопатия боксеров – практически единственная ситуация, когда черепно-мозговые травмы играют роль в развитии П. В целом у людей, не занимающихся боксом, наличию черепно-мозговой трамы в анамнезе (одной или нескольких) не придается этиологическое значение.
П иногда может иметь место при хорее Гентингтона
, которое обычно протекает с выраженными гиперкинезами. Это наследственное заболевание обычно дебютирует на четвертом десятилетии жизни, однако встречаются случаи и более раннего начала. Обычно с акинетико-ригидным синдромом протекает так называемая ювенильная форма хореи Гентингтона (форма Вестфаля). Встречаются также семейные случаи П, в том числе ювенильного. Поэтому у всех больных, имеющих синдром П, сбор наследственного анамнеза имеет большое значение.
Акинезия и ригидность могут иногда наблюдаться при болезни Альцгеймера, Пика,
при которых ядром клинической картины является прогрессирующая деменция.
При болезни Альцгеймера в первую очередь страдает память, а при болезни Пика на первый план выступает распад личности.
При этих заболеваниях П чаще проявляется умеренной акинезией, ригидность выражена меньше, а паркинсонический тремор практически никогда не наблюдается.
Кортико-базальная дегенерация
(КБД) – одна из очень редких причин П. При КБД акинезия и ригидность строго асимметричны, обычно присутствуют в одной руке и сопровождаются миоклониями в ней, дистоническими явлениями и апраксией. Характерный признак болезни –
«синдром чужой руки»
. Последнее проявляется тем, что рука больного независимо от его желания и намерений совершает разные движения, иногда даже достаточно сложные моторные акты.
Постэнцефалитический П
в наше время практически не встречается. Много случаев наблюдалось после пандемии летаргического энцефалита в начале XX столетия. С тех пор в литературе описано только несколько случаев.
Особая эндемическая форма П наблюдается на острове Гуам
и в некоторых других местах восточной части Тихого океана. В этих местах отмечается много случаев П в сочетании с амиотрофиями и деменцией (гуамский комплекс паркинсонизм – деменция). Патоморфологически мозг этих больных выглядит, как мозг больных ПНП. Одинаковыми являются также гистологические маркеры.
Лечение
Людей с МСА лечат препаратами симптоматической терапии. Они помогают убрать выраженность паркинсонизма, мозжечковой атаксии и других признаков дегенерации головного мозга.
В симптоматическое лечение мультисистемной атрофии включают:
- медпрепараты леводопы;
- вазоактивные средства;
- нейрометаболические лекарства;
- массаж, водные процедуры, ЛФК, другие методы физиотерапии;
- диету с соблюдением нормы потребления соли;
- немедикаментозные способы устранения ортостатистической гипотензии.
Паркинсонизм в начальных этапах прогрессии МСА лечат комбинированными средствами леводопы с бенсеразидом, карбидопой. Его заменяют при плохой переносимости или неэффективности препаратами альтернативной терапии. При атрофии всех типов используют агонисты дофаминовых рецепторов, лекарства с веществом амантадин.
Проявления мозжечковой атаксии убирают методами физиотерапии. Для облегчения состояния МСА назначают средства с клоназепамом, габапентином, буспироном, пропранололом и прочими веществами.
При тазовых нарушениях на фоне атрофии используют лекарства:
- Силденафил (при эректильной дисфункции);
- Макрогол, слабительные средства (в случае запоров);
- Антагонист альфа1-адренорецепторов + холинергический препарат (задержка мочи);
- Антихолинергическое средство (непроизвольное или болезненное мочеиспускание).
При нарушениях функций мочевого пузыря дополнительно к лекарствам показана периодическая либо постоянная катетеризация органа. Из альтернативных препаратов применяют уколы ботулотоксина. Эти инъекции также назначают для лечения дыхательных расстройств, каптокормии, слюнотечения, дистонии на фоне МСА.
При ортостатической гипотензии показано спать на кроватях с возвышением изголовья, пить много воды, богатой минералами. Надо носить компрессионные чулки. Нельзя переедать, резко вставать после пробуждения. Рекомендуется обучиться изометрическим маневрам. Прописывают минералокортикоиды, гипертензивные средства с мидодрином.
При нарушениях дыхания показана вентиляция легких либо трахеостомия. Камптокормию лечат методами физиотерапии. Лекарствами с зопиклономили клоназепамом устраняют расстройства сна из-за МСА. При депрессии прописывают селективный ингибитор обратного захвата серотонина (группа антидепрессантов).
Справка! Клиницисты продолжают искать новые методы лечения мультифокальной атрофии. Этиологическую терапию нет возможности применять, пока невыяснены причины и механизм развития МСА.
Осложнения
К последствиям прогрессирования МСА относят урогенитальные инфекции, цистит, уретрит, воспаление почек. Бактерии могут проникнуть в кровь и вызвать сепсис. На фоне дыхательных нарушений чаще развивается пневмония, ночное апноэ.
При мультисистемной атрофии возможно поражение продолговатого мозга. Это осложняется расстройством глотательной функции, вызывает смерть из-за паралича дыхательного центра или асфиксии. Вследствие дегенеративных процессов нарушается также работа сердца, сосудистой системы, мозговое кровообращение.
Прогноз и продолжительность жизни
МСА – неизлечимая патология с необратимыми изменениями мозга. Поэтому прогноз выздоровления негативен. Лечение облегчает проявления мультисистемной атрофии, но не способно остановить или затормозить дегенерацию нейроглии. Качество жизни быстро ухудшается, человек не может себя сам обслуживать.
Смерть наступает в среднем через 7 лет после проявления первых признаков болезни. При вялотекущем прогрессировании продолжительность жизни удваивается. Смерть вызывают осложнения МСА: инфекции, сердечная или дыхательная недостаточность, нарушение мозгового кровообращения.
Прогноз
Лечение исключительно симптоматическое. Основу его составляет леводопа и другие противопаркинсонические препараты
Мультисистемная атрофия – неизлечимое на данный момент заболевание с неуклонно прогрессирующим типом течения. У пациентов быстро ухудшается качество жизни, а через 5–7 лет после дебюта они уже могут оказаться неспособными к самостоятельному передвижению и ведут полупостельный образ жизни.
По мере прогрессирования болезни присоединяются осложнения. Чаще всего отмечаются:
- рецидивирующая урогенитальная инфекция;
- бронхопневмонии на фоне нарастающей дыхательной недостаточности;
- бульбарный синдром с нарушениями глотания;
- апноэ (остановки дыхания) во сне;
- грубые нарушения в работе сердечно-сосудистой системы;
- острые нарушения мозгового кровообращения на фоне эпизодов гипотонии или вследствие ночной гипертонии.
Продолжительность жизни пациентов с мультисистемной атрофией после развертывания основных симптомов невелика, хотя описаны случаи относительной стабилизации состояния на несколько лет. Раннее появление и быстрое нарастание вегетативной недостаточности ухудшают прогноз. Летальность обычно связана с острыми сосудистыми нарушениями, сепсисом, пневмонией, апноэ.
Методы профилактики
Поскольку до конца не изучены причины и патогенез МСА, специфическая профилактика не разработана. Врачи рекомендуют при работе с нейротоксическими веществами пользоваться респираторами и прочими средствами индивидуальной защиты.
Для профилактики генетической предрасположенности беременной женщине категорически запрещено курить, употреблять наркотики, алкоголь, принимать лекарства с тератогенным и эмбриотоксическим свойством. Лекарства назначает только врач по строгим медицинским показаниям.
Что нужно запомнить?
- При мультисистемной атрофии в мозге происходит необратимая дегенерация тканей центральной нервной системы.
- Причины возникновения и механизм развития продолжают исследоваться.
- В справочнике МКБ-10 оставили два типа болезни: паркинсонический и мозжечковый.
- В клинике МСА присутствуют 2―3 синдрома одновременно: вегетативная недостаточность в сочетании с мозжечковой дисфункцией и/или паркинсонизмом.
- В ходе диагностики пациента тестируют на различные виды расстройств ЦНС, делают томографию мозга.
- Лечение мультифокальной атрофии головного мозга заключается в применении симптоматической терапии.
- Сепсис, бульбарный паралич и иные осложнения МСА приводят к смерти.
- Прогноз на выздоровление отсутствует, пациент живет максимум 15 лет.
- Профилактика МСА не разработана, поскольку невыяснены причины болезни.
Литература
- Шиндряева H.H., Белова А.Н., Левин О.С. Мультисистемная атрофия — распространённость в Нижнем Новгороде. Сборник материалов научно-практической конференции «Вегетативные расстройства в клинике нервных и внутренних болезней -2009» 2009 г.
- Шиндряева H.H., Левин О.С. Вегетативные проявления у больных с мультисистемной атрофией. Сборник материалов III научно-практической конференции «Вегетативные расстройства в клинике нервных и внутренних болезней» 2010 г.
- Дамулин И.В., Яхно H.H., Гончаров O.A. Сравнительная оценка нарушений высших мозговых функций при различных типах церебральной атрофии.// Журн. неврологии и психиатрии С.С.Корсакова -1990.
- Пономарев, В.В. Редкие неврологические синдромы и болезни. -СПб. Фолиант.
- Brown RG, Lacomblez L, Landwehrmeyer BG, Bak T, Uttner I, Dubois B, Agid Y, Ludolph A, Bensimon G, Payan C, Leigh NP; for the NNIPPS Study Group (August 2010). “Cognitive impairment in patients with multiple system atrophy and progressive supranuclear palsy”. Brain. 133(Pt 8): 2382—93.
- Swan L, Dupont J (May 1999). “Multiple system atrophy”. Phys Ther. 79(5): 488—94. PMID 10331752.
- American Psychiatric Association. Diagnostic and statistical manual of mental disorders, 4th edition, (DSM IV).// Washington, DC: American Psychiatric Association, 1994.
- The Consensus Committee of the American Autonomic Society and the American Academy of Neurology (1996). “Consensus statement on the definition of orthostatic hypotension, pure autonomic failure, and multiple system atrophy”. Neurology. 46(5): 1470. PMID8628505.