Организм человека состоит из огромного количества клеток. Они в свою очередь объединяются в более крупные единицы – ткани и органы. Из последних строятся системы органов, а и из них и получается организм. И на всех уровнях постоянно идут различные взаимодействия и сложные биохимические реакции. Чтобы все это гармонично развивалось из оплодотворенной яйцеклетки и слаженно функционировало, требуется специальная схема работы для всех уровней. Ее роль в организме выполняет организованная генетическая информация.
Именно на наследственный материал возлагается задача по сохранению данных об общем облике человека, особенностях строения отдельных органов, принципах регуляции при помощи гормонов, и даже того, каким образом должны собираться белки. Все данные записываются в виде последовательности структурных единиц – нуклеотидов. Они подобно буквам алфавита формируют группы-слова, выполняющие определенные функции. Каждая группа называется геном. Все вместе они формируют молекулу ДНК. Если посмотреть на уровень выше, то можно заметить, что весь наследственный материал в виде цепочек дезоксирибонуклеиновой кислоты сконцентрирован в особых образованиях, которые называются хромосомами.
Их у человека в норме насчитывается 23 пары. В каждой паре информация хранится в двух копиях. Это необходимо для того, чтобы при передаче генетической информации каждая дочерняя клетка получила свою копию. Одна пара хромосом отличается от остальных и отвечает за вопросы определения пола. Если в ней имеются две одинаковые большие хромосомы (ХХ вариант), то организм относится к женскому полу. Если же имеется одна большая и одна малая хромосома, то речь идет о мужском поле (XY вариант). Эти две отличающиеся от прочих хромосомы называются половыми. Оставшиеся 22 пары встречаются во всех организмах независимо от пола. Их называют аутосомами.
Аномалии генетического материала
Наследственный материал состоит из огромного количества нуклеотидов, формирующих гены. При этом в каждом гене последовательность нуклеотидов строго определена, поскольку должна кодировать определенный белок. Кроме того, сами гены при формировании хромосом также выстраиваются в фиксированном порядке. Благодаря сохранению этого порядка организм может функционировать, а ученые – быстро и точно указывать друг другу, про какой ген идет речь.
В идеальном случае система работает без малейших сбоев, а генетическая информация всегда передается в неизменном виде. Однако на практике большое число структурных единиц и постоянное воздействие различных факторов (например, ионизирующего излучения) приводит к тому, что время от времени возникают различные аномалии. В частности, отдельные участки последовательности ДНК могут быть скопированы на новое место. В таком случае говорят о дупликации. Если же вместо создания новой копии была перемещена часть исходной цепочки, то модификация называется транслокацией. Кроме того, иногда часть последовательности просто теряется, удаляется из генетического материала. В таком случае изменение называется делецией.
Поскольку взаимодействия в организме оттачивались в течение многих тысячелетий эволюционного развития, получилась очень слаженная система. И аномалии, даже самые небольшие, могут вызвать нарушение баланса. В таком случае в организме развивается то или иное нарушение. Если при этом причина находится на уровне генов, то говорят о генных болезнях. Если была утрачена или наоборот получена лишняя копия хромосомы, то такие нарушения называются хромосомными заболеваниями.
Результаты лечения в Германии
Татьяна (мама пациентки): «Наш результат: Виолетте поставили диагноз синдром Ангельмана (генетическое заболевание, которое подтвердилось также генетическим исслдеованием). Поле начала приема медикамента для сна эпилептические приступы, как ни странно, прекратились, так как дочка начала спать и у нее наладился режим день-ночь.
Второй раз в Германию для подбора медикаментов от эпилепсии мы не полетели, т.к. эпилепсии теперь просто нет. Делали ЭЭГ — всё тихо и спокойно в голове у нашей дочки. Наши врачи сказали, что такого не бывает, чтобы в течение месяца так изменилась энцефалограмма. Результаты ЭЭГ мы с помощью GLORISMED переслали доктору в Берлин – доктор также подтвердила, что эпилептической активности нет.
Столько было в России потрачено сил, нервов, денег… А здесь три дня пребывания в Берлине — и нам рассказали, как дальше жить и бороться с теперь понятным заболеванием!!!
Дорогие родители, не тратьте время и деньги на обследование и терапию в России или других (особенно азиатских) странах. Найдите причину заболевания и уже после этого проводите правильную терапию с правильными докторами. В Германии лучшие врачи и диагносты. Поверьте, когда знаешь, что именно лечить, то лечение дешевле и результативнее!
О компании GLORISMED. Очень благодарна сотрудникам. Рада, что попала именно к ним. Отвечают на все вопросы, организуют и сопровождают во время пребывания в Германии, постоянно на телефоне. Всё делают грамотно и спокойно в любой ситуации.
Даже уже находясь в России, я постоянно задаю сотрудникам вопросы по лекарствам, по связи с доктором и т.д. Ответы получаю быстро, и всё становится понятно. Просто низкий поклон ребятам из GLORISMED, потому что настроение у нас, мамочек, бывает разным, решение нужно здесь и сейчас — и у них обеспечивать это решение получается великолепно!»
Эпилепсия
Эпилепсия. Эпилептология. Лечение детей с эпилепсией. Эпилепсия принадлежит к часто встречающимся неврологическим заболеваниям особого вида, которые угнетают человеческую жизнь
Позвольте помочь Вам найти правильное решение в сложившейся непростой ситуации с Вашим здоровьем.
Свяжитесь с нами любым удобным для Вас способом: по телефону, по электронной почте, по скайпу в режиме онлайн, заполнив контактный формуляр.
Сообщите нам свой контактный номер телефона, наши сотрудники перезвонят Вам и уже сейчас охотно ответят на все Ваши вопросы абсолютно бесплатно и соориентируют Вас по правильному подбору лечебного учреждения, порекомендуют грамотного специалиста – известного врача одной из серьезных профильных немецких клиник, а также назовут Вам приблизительную стоимость лечебной программы, которая требуется именно Вам или близким Вам людям.
Консультация для Вас абсолютно бесплатна!
Что такое микроделеционный синдром?
Самые незначительные изменения (они же мутации) называются точечными. Их появление влияет на считанные единицы генов. В некоторых случаях нарушение относится вообще к одному единственному гену. Однако если он обеспечивал выработку важного белка, последствия для всего организма могут быть очень серьезными. Подобные патологические изменения относятся к группе микроделеционных синдромов.
Каждое такое заболевание обусловлено небольшим изменением генетического материала, которое происходит в строго определенном месте. Точный механизм возникновения подобных нарушений на сегодняшний день не установлен, что не мешает ученым заниматься исследованием их воздействия на организм.
Так, было выяснено, что развитие синдрома в таком случае может происходить несколькими различными вариантами. В частности, ряд заболеваний характеризуется участием онкогенов. В других случаях на воздействие непосредственно самой делеции накладывается эффект хромосомного импринтинга и возможные однородительские дисомии.
Частота возникновения большей части микроделеционных синдромов крайне невелика: порядка 1 случая на 50-100 тысяч новорожденных. Набор клинических признаков обычно выражается отчетливо. Для того чтобы поставить диагноз, бывает достаточно лишь совокупности симптомов. Однако при таком подходе невозможно точно прогнозировать здоровье потомков, поэтому зачастую наряду с проверкой обычных признаков производится молекулярно-генетическая диагностика пробанда и его родственников (обычно родители, в некоторых случаях также требуется анализ генотипа братьев, сестер, теть, дядь и так далее).
Патологические проявления сильно отличаются. В частности, их проявление определяется тем, насколько большой участок генетического материала был утрачен в результате делеции. Кроме того, в ряде случаев играет роль то, от кого из родителей была получена мутация (влияние хромосомного импринтинга). Хорошей иллюстрацией последней ситуации является пара синдромов Прадера-Вилли и Ангельмана. Они оба обусловлены наличием делеции в 15 хромосоме. Однако из-за различного механизма действия при передаче от разных родителей клиническая картина этих заболеваний значительно отличается.
Патогенез и причины
Данный дефект возникает из-за делеции (потери участка генетического материала) сегмента 15 хромосомы. Среди других причин часто называют одноотцовскую дисомию (наследование двух отцовских копий хромосомы, вместо копий от обоих родителей), транслокацию или мутацию одного гена.
Также синдром может появляться как результат мутации гена, участвующего в метаболизме убиквитина.
У большей части пациентов с данной патологией в семейном анамнезе нет генетических отклонений. Однако для определенного процента больных данный синдром является унаследованным. Риски становятся выше, если родители имеют хромосомные аномалии. Интересный факт – если в семье родился ребенок с этим синдромом, то вероятность, рождения еще одного ребенка с такой же патологией, составляет 1%.
Появление синдрома Ангельмана достаточно спонтанно, и практически в любой семье может родиться ребенок с этим заболеванием. Не существует конкретных статистических данных о количестве детей с этим синдромом.
По различным подсчетам количество новорожденных с синдромом куклы колеблется от одного на десять тысяч до одного на двадцать тысяч. Ученые предполагают, что на самом деле это число значительно больше.
Позитивное влияние некоторых делеций на жизнеспособность
Небольшие изменения делеционного характера могут существенно повлиять на выживание организма. К примеру, утрата гена, кодирующего белок CCR5-δ32, становится причиной невосприимчивости к вирусу иммунодефицита человека. Ученые предполагают, что эта мутация впервые появилась около 2,5 тысячелетий назад и с течением времени распространилась по территории Европы.
Имеющееся на сегодняшний день распределение отличается неравномерностью. Согласно статистическим данным, около 10% жителей европейских стран устойчиво к ВИЧ. Вместе с тем в скандинавских государствах этот показатель достигает 14-15 процентов. Русские и финны демонстрируют 16-процентный уровень устойчивости. В то же время для Сардинии частота равняется скромным 4 процентам.
Ряд ученых выдвинул гипотезу, что подобное распространение определяется прошедшими в средневековый период эпидемиями бубонной чумы. Вероятно, мутация в гене вызывает повышенную сопротивляемость этому заболеванию. Поэтому на территории тех стран, где прошлась «черная смерть», выжило больше людей с этим генотипом.
Влияние делеций на способность к оплодотворению
Делеции, происходящие в обычных хромосомах (аутосомах) могут в некоторых случаях быть компенсированы нормальной копией гена. Однако когда речь заходит о половых хромосомах, особенно об Y-хромосоме, ситуация меняется.
Прежде всего необходимо отметить, что локализованные на ней гены не имеют второго экземпляра. При нормальном количестве хромосом в наборе Y-хромосома оказывается крайне уязвимой. В сочетании с малым количеством генов это приводит к серьезным последствиям каждого изменения. Особый интерес представляют мутации, касающиеся AZF-локуса и SRY гена.
Патогенез
В норме человек наследует материнскую и отцовскую копии 15 хромосомы. В важной для развития синдрома Ангельмана части хромосомы процесс преобразования наследственной информации от материнского и отцовского гена в белок или РНК (экспрессия генов) заметно отличается.
Экспрессия данных генов связана с гендерным эпигенетическим импринтингом, при котором преобразование определенных генов зависит от того, какая родительская аллель поступила. Для большинства генов преобразуются обе родительские аллели, но при импринтинге преобразованию подлежит только один родительский аллель (такие гены составляют менее 1%). Признаки, которые определяются данными генами, не наследуются по закону Менделя.
Импринтинг происходит благодаря отсутствию изменений в нуклеотидной последовательности молекулы ДНК при модификации молекулы. Последовательность нуклеотидов ДНК распознается ферментом РНК-полимеразой (осуществляет транскрипцию).
В норме на данном участке 15-й хромосомы преобладает экспрессия аллели матери, а отцовская аллель проявляется слабо. При мутациях или утрате материнской копии генов или дефектах геномного импринтинга развивается синдром Ангельмана.
К развитию синдрома приводит также мутация гена UBE3A, необходимого для метаболизма убиквитина. Этот ген присутствует в обоих родительских хромосомах. Отцовская копия гена инактивируется в гиппокампе и мозжечке, а аллель матери практически всегда сохраняет активность. К появлению синдрома обычно приводит утрата материнской копии хромосомы в регионе 15 q11-13, которая провоцирует отсутствие преобразования последовательности нуклеотидов ДНК родительской копии UBE3A в мозге. Отсутствие экспрессии материнской копии данного гена приводит к серьезным отклонениям в деятельности гиппокампа.
Аномалии гена SRY (Sex-determining Region Y)
Ген SRY, как следует из его названия, отвечает за крайне важную функцию. Именно его наличие в хромосомном наборе запускает процесс формирования организма по мужскому фенотипу и стимулирует развитие соответствующих половых органов.
Наличие даже небольшой делеции в этом гене нарушает механизм дифференцировки пола. В результате при нормальном кариотипе 46XY зародыш начинает развиваться как женский организм. По этой причине на ген SRY приходится наибольшее число мутаций, связанных с неразвитостью гонад. Кроме того, изменения этого гена вызывают инверсию пола.
Подготовка к назначению
Позвоните своему врачу, если ваш ребенок или ребенок не достигли ожидаемых этапов развития или имеют другие симптомы или симптомы, общие для синдрома Ангельмана. Затем ваш врач может обратиться к врачу, который специализируется на состояниях, которые влияют на мозг и нервную систему (невролог).
Вот некоторая информация, которая поможет вам подготовиться к встрече.
Что ты можешь сделать
- Запишите признаки или симптомы, которые вы заметили у своего ребенка, и как долго.
- Принесите детские книги и другие записи о развитии вашего ребенка к назначению. Фотографии и видеозаписи могут быть полезны.
- Перечислите основную медицинскую информацию вашего ребенка,включая другие условия, в которых лечится ваш ребенок, а также названия лекарств, витаминов или добавок, которые он или она принимает.
- Попросите члена семьи или друга присоединиться к вам для назначения вашего ребенка. Если врач вашего ребенка упоминает о возможности развития расстройства, вам может быть трудно сосредоточиться на чем-либо, о чем говорит врач. Возьмите кого-нибудь, кто может предложить эмоциональную поддержку и поможет вам запомнить эту информацию.
- Запишите вопросы, чтобы спросить своего врача.
Вопросы, задаваемые врачом вашего ребенка, включают:
- Что может вызвать признаки и симптомы моего ребенка?
- Существуют ли другие возможные причины этих признаков и симптомов?
- Какие тесты нужен моему ребенку?
- Должен ли мой ребенок видеть специалиста?
Вопросы, задаваемые специалистом, включают:
- У моего ребенка синдром Ангельмана?
- Каковы возможные осложнения этого состояния?
- Какие методы лечения доступны?
- Какое лечение вы рекомендуете?
- Какова долгосрочная перспектива для моего ребенка?
- Должен ли мой ребенок или я пройти тестирование на генетические мутации, связанные с этим заболеванием?
- Какие другие специалисты должен видеть мой ребенок?
- Как я могу найти другие семьи, которые справляются с синдромом Ангельмана?
Не стесняйтесь задавать и другие вопросы.
Что ожидать от вашего врача
Врач, который видит вашего ребенка в возможном синдроме Англмана, скорее всего, задаст вам ряд вопросов, таких как:
- Каковы признаки и симптомы вашего ребенка и когда вы их заметили?
- У вашего ребенка проблемы с кормлением?
- Ваш ребенок достигает ожидаемых, связанных с возрастом физических вех?
- Вы заметили проблемы с балансом, координацией или движением?
- Ваш ребенок смеется, улыбается или выражает волнение чаще, чем его или ее сверстники?
- Ваш ребенок выражает волнение с необычным физическим поведением, таким как ручка?
- Сообщает ли ваш ребенок устно?
- Насколько хорошо спит ваш ребенок?
- У вашего ребенка были судороги? Если да, то как часто?
- Были ли у кого-либо из родственников первой степени вашего ребенка, например родителя или брата, был диагностирован синдром Ангельмана?
Поделиться ссылкой:
- Нажмите, чтобы поделиться на Twitter (Открывается в новом окне)
- Нажмите здесь, чтобы поделиться контентом на Facebook. (Открывается в новом окне)
- Нажмите, чтобы поделиться в Telegram (Открывается в новом окне)
Понравилось это:
Нравится
Похожее
Аномалии AZF-локуса
На Y-хромосоме также имеется особый участок, который контролирует процесс выработки сперматозоидов. Именно от этого зависит, насколько эффективным будет сперматогенез. Кроме того, состояние этого участка сказывается на свойствах сперматозоидов, таких как общее количество в эякуляте, способность двигаться, наличие структурных изменений и способность к оплодотворению. Только при наличии хорошо сформированных подвижных сперматозоидов мужской генетический материал может быть доставлен до яйцеклетки. Иными словами, о состоянии этого небольшого участка генетического кода зависит способность мужчины иметь детей.
При наличии в AZF-локусе аномалий процесс выработки сперматозоидов нарушается. В результате могут развиться азооспермия и олигозооспермия. При этих патологиях в эякуляте либо совсем не содержится сперматозоидов, либо их число сильно снижено.
Сам AZF локус делится на три части со специфическими задачами. Они именуются путем добавления индекса: AZFa, AZFb и AZFc. Возникшая делеция может удалять фрагмент отдельной части, либо ее целиком, либо захватывать сразу два региона. При полном удалении AZF развивается тяжелое поражение сперматогенеза. Частичные делеции могут проявляться по-разному. При этом на степень проявления патологии влияют размеры утраченного фрагмента и его расположение в локусе. Поэтому для прогностических целей крайне важно знать, в каком месте произошла делеция. Кроме того, эта информация может использоваться для правильного планирования семьи и проведения экстракорпорального оплодотворения.
Если при делеции был удален весь локус или любой из регионов с индексами a/b, то у мужчины не могут быть получены жизнеспособные сперматозоиды. Если делецию можно описать формулой AZFb/AZFb+, то развивается азооспермия из-за тяжелых нарушений процесса формирования сперматозоидов.
Делеции участка AZFc приводят к проявлению патологических симптомов различной степени тяжести. В том числе возможно развитие олигоспермии, которая в принципе допускает зачатие. В 50-70 процентах от общего числа подобных случаев возможно получение сперматозоидов для дальнейшего использования в методах искусственного оплодотворения. Частичная делеция региона AZFс может выражаться в форме различных нарушений от нормозооспермии до азооспермии.
Все делеции в AZF-локусе, вызывающие ту или иную патологическую ситуацию, являются причинами мужского бесплодия. Определение мутации возможно путем гистологического анализа семенной жидкости. При этом необходима остановка созревания сперматозоидов или обнаружение незрелых сперматозоидов. Для получения точных данных о делециях в AZF-локусе используется ПЦР 6 маркеров, которые относятся к отдельным участкам локуса.
Синдром Ангельмана
При синдроме Ангельмана развивается характерный набор патологических изменений. В частности, отмечается задержка психологического развития, сопровождающаяся проблемами со сном, частыми хаотическими движениям (больше руками), постоянными улыбками и смехом.
Патология развивается при отсутствии некоторых генов, расположенных на 15 хромосоме. При этом обязательным условием является передача мутантной копии гена от матери. Если поврежденная хромосома будет унаследована от отца, то разовьется синдром Прадера-Вилли. Кариотип обычно нормальный (46XX и 46XY для девочек и мальчиков соответственно). Различные независимые исследования указывают на связь болезни с геном UBE3A, который в норме обеспечивает выработку ферментного компонента в сложной системе деградации белков.
Частота появление синдрома составляет примерно 1 случай на 10-20 тысяч новорожденных (показатели отличаются у различных ученых).
Характерными особенностями больных с синдромом Ангельмана являются следующие признаки:
· проблемы с питанием, начинающиеся еще во время грудного вскармливания, поскольку дети плохо набирают вес (распространенность признака порядка 75 процентов);
· заторможенное развитие навыков общей моторики, то есть дети позже других начинают сидеть и ходить;
· для всех детей характерны нарушения речевого развития;
· больные обычно понимают больше, чем в состоянии выразить при помощи ограниченного словарного запаса;
· часто заболевание сопровождается дефицитом внимания и гиперактивностью;
· проблемы с обучением в обычной школе;
· у 80% заболевших развивается эпилепсия, сопровождающаяся заметными на электроэнцефалографии нарушениями; ученые полагают, что заболевание эпилепсией носит вторичный (симптоматический) характер.
· выполнение необычных движений, к которым относятся произвольные хаотические движения конечностями, мелкий тремор;
· возникновение приступов смеха при отсутствии видимых причин;
· характерная ходьба на негнущихся ногах, из-за которой возникло сравнение с марионетками;
· уменьшенная по сравнению со средними размерами голова, часто с уплощенным затылком;
· в некоторых случаях встречаются своеобразные запоминающиеся черты лица – широкий рот с редко расположенными зубами, выдвинутый вперед подбородок с выпущенным наружу языком;
· различные нарушения сна;
· примерно в 40 процентах случаев развивается косоглазие;
· порядка 10% больных также страдает от искривления позвоночника;
· высокие температуры воспринимаются с повышенной чувствительностью;
· наибольшего комфорта обычно достигают в воде (к примеру, в ванной)
Как правило, синдром определяется при помощи методов молекулярно-генетической диагностики по 15 хромосоме. Показанием к проведению тестирования для новорожденного является пониженный мышечный тонус (гипотонус), заметное отставание в развитии речи и мелкой моторики. Кроме того, на заболевание могут указывать мелкий тремор, порывистые беспорядочные движения, передвижение на негнущихся ногах.
Анализ может проводиться через флуоресцентную гибридизацию in situ, метилированием ДНК в области 15q11-q13. Также можно проверить мутации в импринтинговом центре и в гене UBE3A.
Поскольку заболевание обусловлено генетическим нарушением, адекватного и действенного способа лечения для него не имеется. Выполнение лечебных мероприятий, таких, как массаж для больных с гипотонусом, позволяет повысить качество жизни.
лечение
Не существует лечения синдрома Ангельмана. Исследование фокусируется на нацеливании определенных генов на лечение. Текущее лечение сосредоточено на решении медицинских и проблем развития.
Команда медицинских работников, вероятно, будет работать с вами, чтобы управлять состоянием вашего ребенка. В зависимости от признаков и симптомов вашего ребенка лечение синдрома Ангельмана может включать:
- Антиадгезивный препарат для борьбы с судорогами
- Физическая терапия, чтобы помочь с ходьбой и проблемами движения
- Коммуникационная терапия, которая может включать в себя язык жестов и передачу изображения
- Поведенческая терапия, чтобы помочь преодолеть гиперактивность и короткий промежуток внимания и помочь в развитии
Синдром Прадера-Вилли
Это заболевание определяется той же самой генетической мутацией, что и для синдрома Ангельмана. Отличие состоит в том, что при этом нарушение наследственного материала получается со стороны отца. Кариотип соответствует нормальному (46XX или 46XY). По распространенности (1 случай на 12-15 тысяч новорожденных) примерно совпадает с распространенностью синдрома Ангельмана.
Характерными признаками синдрома Прадера-Вилли являются следующие симптомы:
· в пренатальный период малая подвижность плода;
· часто встречается неверное положение плода;
· возможна дисплазия тазобедреных суставов;
· к двум годам может проявиться склонность много есть (больше нормы), что приводит к ожирению;
· низкий мышечный тонус (гипотонус), сочетающийся с нарушенной координацией движений;
· стопы и кисти обычно маленькие, кроме того характерен невысокий рост;
· формирование косоглазия и сколиоза;
· отмечают повышенную сонливость;
· плотность костей находится на более низком уровне, чем у здоровых людей;
· слюна густая, обычно состояние зубов плохое;
· недостаточная функция половых желез, вызывающая в итоге бесплодие;
· позднее по сравнению со сверстниками половое созревание;
· больные позже учатся говорить, отстают в психическом развитии;
· внешние признаки включают выраженную переносицу, узкий и высокий лоб, миндалевидную форму глаз, узкие губы.
В большинстве случаев у человека с мутацией насчитывается от одного до пяти признаков заболевания.
Диагностика заболевания проводится путем молекулярно-генетического тестирования, на которое направляются дети с пониженным мышечным тонусом. Зачастую вместо верного диагноза определяется более распространенный «синдром Дауна». Опытный генетик, достаточно часто встречающийся с проявлениями синдрома Прадера-Вилли способен диагностировать его по комплексу внешних признаков.
Синдром лиссэнцефалии Миллера — Дикера
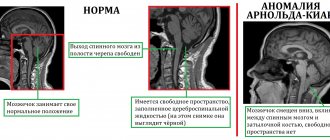
При синдроме лиссэенцефалии Миллера – Дикера причиной патологических изменений является делеция некоторых генов в локусе 17p13. При этом больше всего страдает центральная нервная система. Наряду с лиссенцефалией (сглаживание находящихся на поверхности мозга извилин из-за нарушения деятельности гена PAFAH1B1) отмечается сокращение числа кортикальных слоев. Если в норме их насчитывается 6 штук, то у больных можно обнаружить только 4. Сопутствующими признаками является заметное изменение форм лица. Кроме того, больные медленно растут. Попытки интеграции в общество осложняются множественными патологиями сердца, желудочно-кишечного тракта, почек. Если при заболевании происходит делеция гена 14-3-3 эпсилон, то синдром проявляется значительно тяжелее.
Аниридия
При аниридии нарушается нормальное строение глаза: в органе зрения отсутствует радужная оболочка. Кроме того, часто развиваются сопутствующие патологические изменения, такие как макулярная гипоплазия и гипоплазия зрительного нерва, изменения роговицы, катаракта. Острота зрения заметно падает, попытки коррекции не приносят существенных результатов. Развивается светобоязнь и горизонтальный нистагм. В некоторых случаях отмечается появление врожденной глаукомы.
Причиной заболевания является нарушение функционирования гена PAX6 из короткого плеча 11 хромосомы. Кодируемый им белок приводит к запуску ряда процессов, которые управляют процессом правильного формирования органов зрения и ряда других структур. Примечательно, что ген очень консервативен: отличие форм PAX6 у человека и данио рерио составляет менее 5%, несмотря на расхождение эволюционных линий примерно 400 млн лет назад.
Заболевание относится к группе аутосомно-доминантных патологий. В случае гомозиготности по мутантной копии гена PAX6 негативный эффект на организм возрастает, что вызывает множественные нарушения в работе органов зрения. Кроме того, поражается ЦНС, что приводит к летальному исходу.
Лечение направлено на сглаживание симптомов. Для визуальной имитации зрачка рекомендуется использовать специальным образом окрашенные линзы. Возможно восстановление зрачка путем реконструктивной пластической операции.
Синдром Ди Джорджи
При синдроме Ди Джорджи у больных отмечается наличие врожденной формы аплазии паращитовидных желез и тимуса. Является разновидностью идиопатического изолированного гипопаратиреоза. Встречается достаточно редко.
При этом заболевании патологические изменения касаются околощитовидных (паращитовидных) желез, у которых отмечается дисгенез или агенезия. Вилочковая железа (тимус) отсутствует от рождения. В результате сочетания таких патологий происходит резкое снижение числа Т-лимфоцитов, формируется иммунологическая недостаточность. Кроме того, этот синдром сопровождается формированием врожденных аномалий крупных сосудов.
Заболевание является аутосомным и определяется наличие мутации в 22 хромосоме. В большинстве случаев причиной является спорадическая делеция 22q11 (реже микроделеция 22q11.2). Наследование происходит по доминантному принципу, с полом не связано. Некоторые авторы не соглашаются с такой характеристикой и приводят аргументы в пользу аутосомно-рецессивного типа, обладающего различной эспрессивностью.
Для заболевания характерно нарушение процесса эмбриогенеза 3-4 жаберных карманов, что приводит к нарушению закладки вилочковой железы и паращитовидных желез.
В клинике наиболее постоянными симптомами являются кандидомикоз и гипопаратиреоз, довольно часто сопровождающиеся нарушением процесса формирования рта, носа и ушей.
Тимус из-за нарушения развития в эмбриональном периоде остается неразвитым. Эпителий тимуса не обеспечивает нормального процесса развития Т-клеток. В итоге формируется специфическая форма иммунодефицита, при которой ослабляется гуморальный иммунный ответ и ответ на клеточном уровне. Если у ребенка имеется подобное патологическое нарушение иммунитета, то он будет обладать повышенной чувствительностью к инфекциям бактериального, вирусного и грибкового происхождения.
Синдром может протекать в форме генетически обусловленного отсутствия паращитовидных желез или изолированной недостаточности околощитовидных желез – в сопровождении гипокальциемических судорог, которые начинаются от рождения. Иммунологическая недостаточность приводит к появлению различных инфекционных заболеваний. Как правило, совокупность симптомов вызывает сердечную недостаточность. Кроме того, летальный исход вызывают инфекционные болезни.
Диагностика синдрома предполагает выявление типичных для синдрома патологий: искажения формы лица и черепа, наличие иммунологической недостаточности, аплазии тимуса, дисгенезии или агенезии паращитовидных желез. Ярче всего при заболевании проявляются кандидомикоз и гипопаратиреоз.
Причины развития
Причины синдрома Ангельмана – отсутствие некоторых генов 15-й хромосомы. Данный синдром возникает при мутации материнской хромосомы (дефекты отцовской хромосомы вызывают синдром Прадера – Вили).
В большинстве случаев заболевание связано с утратой (делецией) части хромосомы.
Причиной заболевания может быть спонтанно возникший дефект 15-й хромосомы, который связан с отсутствием в регионе q11—q13 состоящей из 3-4 млн. пар оснований ДНК большой смежной области.
Мутация гена UBE3A также способна вызывать синдром Ангельмана.
Ретинобластома
Ретинобластомой называют злокачественную опухоль сетчатки глаза. Процесс развития начинается обычно в детском возрасте, причем исходным материалом являются ткани эмбрионального происхождения. Пиковая фаза приходится на двухлетний возраст.
Практически все известные случаи выявляются в течение первых 5 лет жизни.
Причиной заболевания в большинстве случаев является мутация в генетическом материале. При этом необходимо наличие генетической обусловленности за счет наличия мутантной версии гена Rb, полученной по наследству. Вторая мутация, вызывающая появление опухоли, происходит в ретинобласте.
Существует небольшая вероятность, что у родителей, которые переболели ретинобластомой, могут родиться дети с отсутствием патологического изменения.
Отмечаются односторонние и двусторонние случаи ретинобластомы. По статистике для двусторонней формы вероятность наследственного происхождения заметно выше.
Симптомы заболевания включают боль в глазах, свечение зрачка, а также потерю зрения. Выявить их у маленького ребенка очень и очень трудно.
Диагностика обычно проходит в форме обследования под наркозом с применением УЗИ, КТ и МРТ. Достаточно распространенным приемом является биопсия красного костного мозга и спинномозговая пункция. По тяжести симптомов выделяется 5 групп.
Существует два эффективных метода лечения. При криотерапии и фотокоагуляции остается возможность сохранить и зрение, и сам глаз. Осложнения при их использовании возникают редко. Тем не менее, если возникнет рецидив, лечение потребуется повторить в той же форме. Обычно криотерапия используется в случаях, когда поврежден передний отдел сетчатки. Для заднего отдела более предпочтительным вариантом представляется фотокоагуляция.
Приезд в Германию на диагностику эпилепсии
Татьяна (мама пациентки): «Спустя всего 10 дней после нашего первого обращения мы прилетели в Берлин. На следующий день после прилета мы поехали в клинику. Там встретились с переводчиком — очень приветливая девушка.
У доктора мы провели около 3 часов. Она посмотрела МРТ, понаблюдала за поведением ребенка, подробно расспросила об истории жизни и болезни — от вынашивания беременности до сегодняшнего дня с уточнением всех подробностей (даже самых малейших), также нам сделали ЭЭГ (сам процесс тоже очень отличается от того, что мы делали в России, в гораздо лучшую сторону).
Доктор сразу нам дала понять, чего примерно мы можем ожидать в жизни Виолетты. И уже через два дня нас позвали для подробного анализа состояния ребенка в данный период и разъяснения информации по диагнозу. Диагноз был предварительный, так как еще требовалось генетическое обследование (которое мы сделали уже в России).
Мы, честно, были шокированы, когда нам озвучили предварительный диагноз: синдром Ангельмана и эпилепсия. Для сравнения: в России врачи (их было много и в разных городах) твердили на протяжении 4 лет, что у нас нетипичный случай ДЦП (формы ставили разные!), эпилепсия, мол, не такая как у всех (откровенно не говорили, но мы понимали по взглядам, что просто не знают, как лечить). А в Германии после трех часов общения и потом просмотра анализов и обследований, сделанных в течение дня (в общем ушло по времени 2 дня), поставили точный диагноз!!!
Для нас это всё было шоком. Огромная благодарность доктору и переводчикам GLORISMED за правильные, понятные переводы — это тоже очень важно (говорю по собственному опыту, т.к. была в Китае и не понаслышке знаю, как переводят там)! В клинике Германии выписали лекарство для сна и просто для нормального самочувствия ребенка.
Для подбора медикаментов от эпилепсии мы хотели приехать позже на более долгий срок. В связи с тем, что на терапию в России у Виолетты была очень тяжелая реакция, подбор медикаментов должен был проходить под наблюдением немецких специалистов».